

Попытки использования генетических и клеточных технологий в медицинских целях для лечения или профилактики заболеваний насчитывают не одно десятилетие и стали предприниматься параллельно с созданием технологии получения рекомбинантных белков. В частности, первые указания FDA (U.S. Food and Drug Administration), делающие попытку упорядочить подходы к разработке таких продуктов и сформулировать критерии для выведения их на рынок, были приняты еще в далеком 1991 г. Вместе с тем первый препарат для клеточной терапии был одобрен в США только в 2010-м (аутологичная клеточная иммунотерапия бессимптомного или минимально симптомного метастатического гормон-резистентного рака предстательной железы — сипулейцел-T), а первый генотерапевтический препарат вышел на рынок США и вовсе в 2015 г. с целью местной терапии нерезектабельных кожных, подкожных и узловатых очагов у пациентов с меланомой, рецидивирующей после радикальной хирургии.
Вот и получается, что, несмотря на достаточно длительную историю разработки этих технологий, конкретная медицинская практика и ее первые плоды появились совсем недавно, а потому генные и клеточные терапии — всё еще очень новое лечение. Такой «неторопливый» старт показывает, что пока наших знаний и умений недостаточно для уверенного создания терапевтических продуктов, лечащих за счет модификации генетического аппарата или восполнения утраченных клеток. Сегодня вклад в медицину этой группы лекарств весьма скромен. Число одобренных продуктов не превышает нескольких десятков, а сами они предназначены для лечения, как правило, редких заболеваний. Но темп разработок растет, и многие заболевания уже находятся «на очереди» у генных и клеточных технологий, а потому нет основания не верить, что в итоге прилагаемые усилия увенчаются успехом.
Недостаточно разработать и протестировать эффективные и безопасные лекарственные препараты генной и клеточной терапий. Разработчикам также нужно организовать их производство и обеспечить всесторонний контроль качества технологических процессов. При этом надо максимально стандартизировать все процедуры, чтобы все производимые серии препаратов были идентичны по своим характеристикам.
Выработкой требований к лекарственным препаратам и критериев их оценки занимается регуляторная наука. Это целый комплекс научных дисциплин, которые имеют отношение к оценке качества, безопасности и эффективности препаратов и создают основу для принятия регуляторных решений на протяжении всего жизненного цикла лекарства. Регуляторная наука охватывает фундаментальные и прикладные медицинские и социальные науки, а также вносит вклад в разработку регуляторных стандартов и инструментов.
В связи с тем, что регуляция лекарств наиболее развита на территориях ICH (International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, Международного совета по гармонизации технических требований к лекарствам для медицинского применения, поддерживающего систему руководств по научно обоснованной разработке лекарств), в первую очередь в США и ЕС, далее мы рассматриваем правила регулирования на основе того, как они изложены в руководствах ICH, а российских особенностей коснемся в последнем разделе.ЖИЗНЕННЫЙ ЦИКЛ ЛЕКАРСТВАсовокупность всех фаз жизни препарата от начальной разработки до воплощения и окончательного вывода с рынка.
Принадлежность к биопрепаратам
Как «Биомолекула» уже писала раньше, историю новых лекарственных разработок удобно рассматривать как идущие друг за другом три поколения лекарств. Первое — низкомолекулярные лекарства, второе — биопрепараты, а третье — передовая терапия, в первую очередь генная и клеточная.
С регуляторной точки зрения генные и клеточные терапии — это лекарства, получаемые из биологических источников [2–4]. И те, и другие — макромолекулярные сущности с чрезвычайно сложными характеристиками, которые невозможно описать и установить полностью. Они производятся биологическими системами (включая организм человека в случае клеточных препаратов), что вносит большую вариабельность в результат процесса производства (об этой особенности биопрепаратов говорилось в материале «Биомолекулы» «Контроль качества биотехнологических продуктов» [5]). В этом ключе в отношении биопрепаратов еще на заре биотехнологической эры возникло понимание, метко описанное высказыванием, что процесс — есть продукт (process is the biologic) [6]. Другими словами, специалисты осознали, что характеристики получаемого биопрепарата (то есть его качество), определяющие профиль его безопасности и эффективности, неотделимы от условий и параметров его производства. Важное следствие: чтобы получить качественный продукт, необходимо строго контролировать параметры производства, потому что их вариабельность, скорее всего, приведет к отклонению в их эффективности и безопасности [7]. Этот факт имел и продолжает иметь конкретный практический аспект: если параметры процесса производства сильно влияют на качество получаемого продукта, то контроль параметров производства вносит существенный вклад в общий профиль качества, а вариабельность таких условий может приводить к получению продукта с другими характеристиками, а по сути, нового продукта.
Эти особенности биопрепаратов определяют [6], [8], [9]:
- высокую специфичность механизма действия и «прицельность», что обусловливает их низкую неспецифическую токсичность, а их небезопасность в первую очередь опосредуется фармакологическим действием (то есть воздействием на мишень [10]) и иммуногенностью, а также способностью к неконтролируемой репликации или пролиферации и росту;
- высокую чувствительность к окружающим факторам, в результате чего как производство, так и хранение, транспортировка, реализация и применение биопрепаратов должны осуществляться в щадящих, строго контролируемых условиях. Клетки-продуценты очень требовательны к условиям содержания;
- неустойчивость во внешней среде, из-за чего биопрепараты, как правило, должны находиться в водных средах или подвергаться лиофилизации, в то время как низкомолекулярные вещества не имеют таких сильных ограничений, и зачастую могут находиться в капсулах, таблетках и т. п.;
- преимущественно парентеральный путь введения (как правило, внутривенно); поскольку при этом препарат поступает во внутренние среды организма, минуя защитные барьеры, их необходимо тщательно стерилизовать, что создает дополнительные проблемы, так как биопрепараты подвержены биологической контаминации и не могут подвергаться агрессивным методам стерилизации и очистки для устранения бионагрузки;
- основной упор на разработку биологического действующего вещества, обеспечение однородности его свойств, очистку и поддержание стабильности; разработка формуляции (водного раствора) относительно проста по сравнению с усилиями, прилагаемыми к разработке лекарственной формы для низкомолекулярных действующих веществ (таблетки, мази, аэрозоли, пластыри).
В то время как для лекарств, содержащих низкомолекулярные действующие вещества, качество нередко можно обеспечить с помощью выборочных испытаний конечного продукта, для биопрепаратов это невозможно. Поэтому определение биопрепарата в Европейском [3] и в Евразийском экономическом [4] союзах выглядит так:БИОПРЕПАРАТлекарство, действующее вещество которого вырабатывается биологическим источником или выделяется из него, требующее в целях установления его характеристик и определения его качества комбинации физико-химических и биологических испытаний вместе с испытаниями процесса производства и его контроля.
Биопрепараты делятся на:
- биотехнологические;
- иммунологические (вакцины, гетерологичные иммуноглобулины и иммуносыворотки, диагностикумы и аллергены);
- препараты, получаемые из плазмы (альбумин, факторы свертывания, иммуноглобулин человека);
- препараты передовой терапии, включающие препараты для генной и клеточной терапий, а также препараты тканевой инженерии;
- нерекомбинантные препараты (ботулотоксины, пищеварительные ферменты, урофоллитропины, гепарины и т. д).
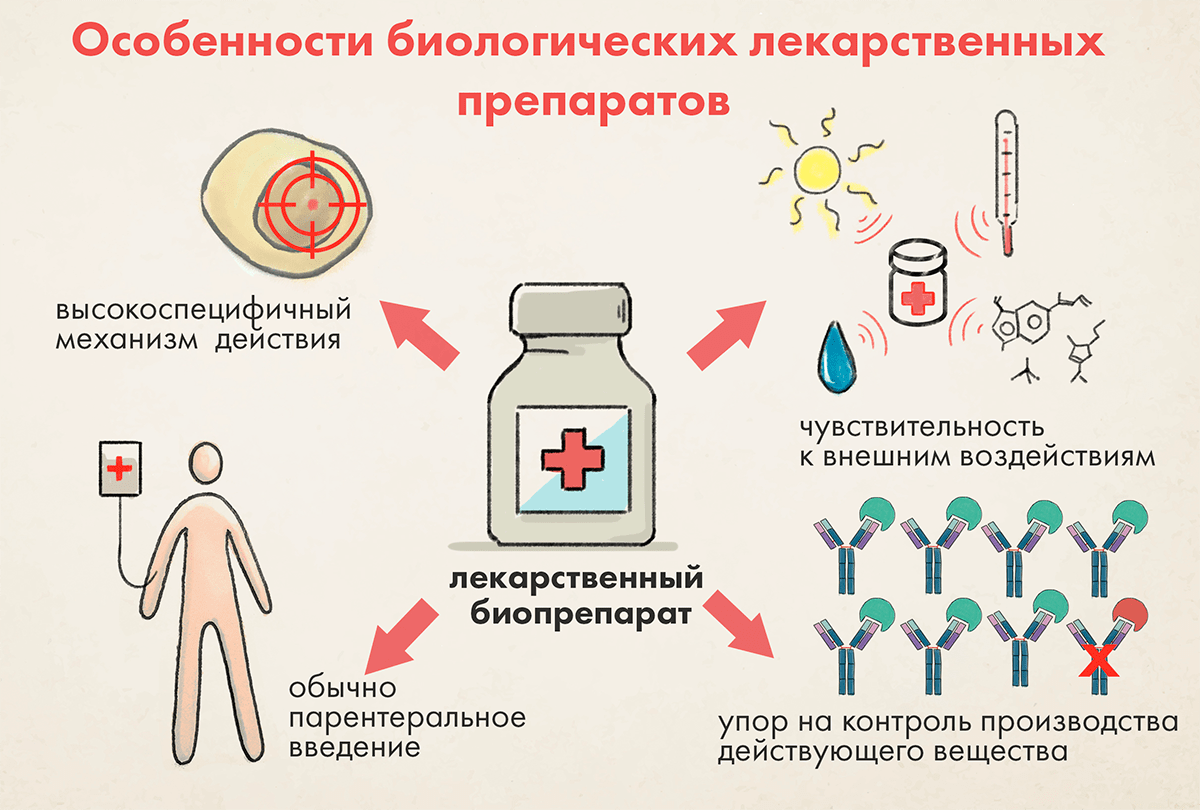
Всесторонний контроль качества не ограничивается тестированием качества биопрепарата по окончании всего процесса производства, а состоит из следующих аспектов (рис. 1):
- тщательный отбор и тестирование исходных и сырьевых материалов, прежде чем они будут введены в процесс производства;
- микробиологический контроль исходных и сырьевых материалов, соблюдение правил асептики во время производства и удаление и инактивация вирусов;
- сложная стратегия контроля многостадийного процесса производства — более сложного, чем синтез низкомолекулярных действующих веществ (табл. 1).
| Требования из расчета на серию АФИ* | Низкомолекулярное вещество | Биопрепарат |
|---|---|---|
| Записи о серии | <10 | >250 |
| Испытания на качество препарата | <100 | >2000 |
| Критичные стадии процесса | <100 | >5000 |
| Число фиксируемых показателей/параметров процесса | <4000 | >60 000 |
| * АФИ — активный фармацевтический ингредиент, компонент лекарственного препарата, являющийся действующим веществом или становящийся им в организме человека. |
Из таблицы видно, насколько биопроцессы сложнее процессов химического синтеза низкомолекулярных веществ. Более того, чем более сложную организацию имеет производимое биологическое вещество, тем труднее будет создать, поддерживать и контролировать этот процесс. А генная и клеточная терапии — это на сегодняшний день и есть самые сложные в производстве биологические препараты.
Хотя у биологических лекарственных препаратов есть общие черты, отличающие их от низкомолекулярных веществ, они представляют собой достаточно неоднородную группу. Разные биопрепараты могут существенно различаться между собой как по характеристикам безопасности и эффективности, так и с точки зрения производства и контроля качества.
Рассматриваемые нами препараты передовой терапии хотя и объединены в общую категорию, также сильно отличаются друг от друга, пересекаясь только в случае терапевтических генетически модифицированных клеток (например, CAR-T [12]; свежий пример одобренного FDA препарата CAR-T — препарат для лечения мантийноклеточной лимфомы брексукаптагена аутолейцел, Tecartus), которым присущи особенности как генотерапевтических конструкций, так и клеточных препаратов. Тем не менее важно рассмотреть отличия передовых терапий от наиболее обширной и развитой группы биопрепаратов — рекомбинантных терапевтических белков. В таблице 2 приведены важные различия в производстве рекомбинантных белков и препаратов передовой терапии, которые приводят к соответствующим рискам для пациента, а также определяют стратегию контроля для их минимизации. Кроме того, особенности производства и контроля также влияют на затраты и тем самым на доступность лекарств для здравоохранения.
| Рекомбинантные белки | Генотерапевтики | Клеточные препараты | |
|---|---|---|---|
| Разработка препарата | Генная инженерия | Генная инженерия | Человеческий материал (иногда в сочетании с генной инженерией) |
| Физическая доступность исходных материалов | Высокая | Высокая | Низкая в случае аутологичных препаратов |
| Источник | Система банков клеток | Система или системы банков клеток (экспрессирующая конструкция, пакующие клетки) | Сами клетки человека (аутологичные препараты) или банк клеток (аллогенные) |
| Клональность | Да | Да | Может быть в случае аллогенных препаратов |
| Возможность установления физико-химических и биологических характеристик | Очень высокая | Умеренная | Сильно ограниченная |
| Очистка | Высокая | Средняя | Почти отсутствует |
| Удаление / инактивация вирусов | Да | Ограниченно | Нет |
| Микробиологические риски | Ничтожны | В случае вирусных векторов — из самого вектора. Есть риск того, что при производстве аденовирусного препарата останутся не инактивированные вирусные частицы | Высокие |
| Выпускающий контроль до введения пациенту | Полный | Полный | Неполный (например, мы не в состоянии, провести формальные микробиологические тесты для CAR-T) |
| Хранение | Длительное | Длительное | Хранение клеток в жидком азоте практически не ограничено по времени, но лимитируется регуляторикой, однако не все клетки хранятся таким образом, для некоторых хранение сверхкороткое или невозможно |
| Трудоемкость и сложность процесса в расчете на дозу | + | ++ | +++, особенно для аутологичных препаратов |
| Основной риск примесей | Иммуногенность | Иммуногенность, вирулентность | Туморогенность, эктопическое биораспределение |
Препараты на основе клеток
Терапевтические продукты на основе клеток достаточно разнообразны:
- живые донорские органы человека, предназначенные для трансплантации;
- живые донорские ткани и клетки человека, предназначенные для трансплантации без какой-либо модификации;
- живые форменные элементы крови человека для трансфузий, не подвергающиеся какой-либо модификации;
- живые клетки человека, включая форменные элементы крови, подвергающиеся различным манипуляциям для придания им определенных свойств или наращивания биомассы;
- живые органы, ткани и клетки животных для ксенотрансплантации.
В нашей статье мы не будем рассматривать ксеногенные материалы и трансплантацию органов и остановимся только на продуктах человеческого происхождения. В качестве лекарственных препаратов регулируются только те клетки человека, которые подвергаются существенным манипуляциям для наделения их полезными свойствами (например, генетической модификации, обработке факторами роста и т. д.) или наращивания биомассы [3], [13]. Регулирование в качестве лекарственных препаратов — самое строгое среди всех потребительских товаров и предполагает выполнение обширных требований, направленных на обеспечение не только их безопасности для пациентов, но и обладание ими заявляемых терапевтических свойств (эффективности) [14].
Отправная точка производства клеточных препаратов — клетки человека (рис. 2). Их получают от живых или посмертных доноров.
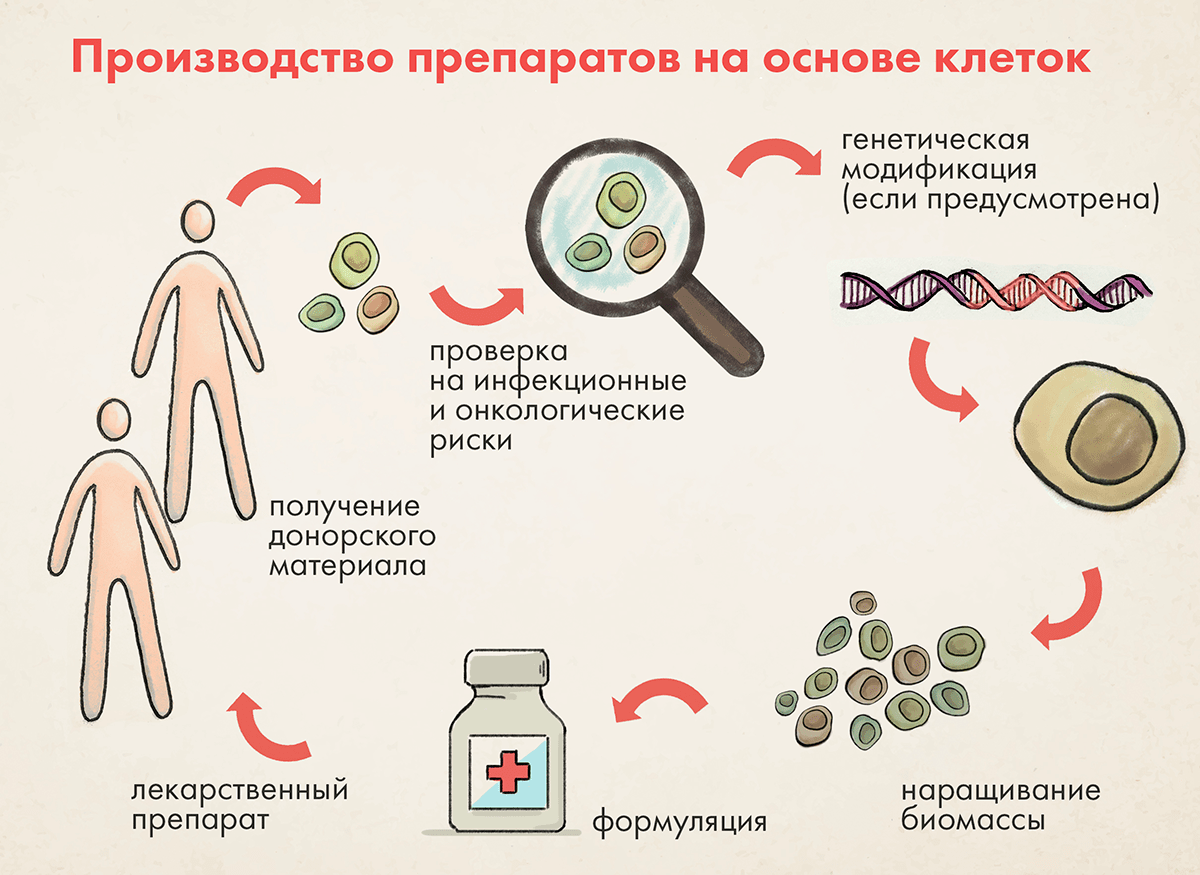
Живые клетки человека могут быть опасны, поэтому в ходе фармацевтической разработки необходимо решать следующие проблемы или минимизировать связанные с ними риски [15–19]:
- Инфекционные и онкологические риски. Донор может быть инфицирован известными или неизвестными микроорганизмами (наибольший риск представляют вирусы и прионы), либо донорский материал может содержать клетки, подвергшиеся или стоящие на пороге опухолевой трансформации.
Эта категория рисков значима, если донорский материал будет использоваться для получения аллогенных препаратов, то есть их реципиентами будут другие люди, а не сам донор. Из-за высокой восприимчивости клеточных препаратов к любым методам деконтаминации (которые по определению агрессивны) для обеспечения микробиологической безопасности невозможна ни химическая, ни термическая, ни лучевая обработка. Кроме того, крупные размеры клеток не позволяют использовать противомикробные фильтры для фильтрации бактерий, микоплазмы и вирусов. В этом ключе главное средство инфекционной безопасности на этапе производства — строжайший отбор доноров. Его критерии могут быть смягчены только в случае аутологичного донорства (то есть донорства для собственных нужд) [20], [21]. Вместе с тем никакой строгий отбор не способен гарантировать полную инфекционную безопасность, поэтому остаточный риск неустраним и требует понимания со стороны лечащего врача и самого пациента и явного принятия этого риска (зачастую для снижения риска на этапе применения используют антибиотики). При несоблюдении правил надлежащей производственной практики (GMP) нередки инфекционные осложнения [22]. - Ограниченный объем донорского материала. Данный аспект особо значим в случае аутологичного донорства, когда клетки заготавливаются для собственных нужд.
Малые количества клеток требуют особо бережного и продуманного выполнения всех производственных процессов, ведь любые серьезные проблемы могут привести к порче получаемого продукта и его отбраковке. Если у пациента-донора имеется дефицит клеточного материала (ведь нередко он решает прибегнуть к клеточной терапии именно для восполнения утраченных клеток), то утрата произведенного продукта может вообще оказаться трагичной. Дефицит донорского материала также накладывает ограничения на контроль качества, поскольку ряд испытаний требует расходования клеточного препарата. Если материала мало, контроль качества может оказаться неполным, а это, в свою очередь, влечет за собой повышенные требования к валидации процесса производства — нередко ключевого компонента обеспечения качества клеточного препарата [15]. - Требовательность к условиям обработки. Клетки очень чувствительны как к окружающим производственным условиям, так и методам обработки.
Будучи живыми, клетки очень восприимчивы к любым неблагоприятным воздействиям в процессе производства. Производство клеточного препарата — это комплекс манипуляций с исходным донорским материалом для придания ему желаемых новых биологических свойств или для его наращивания, чтобы получить достаточную дозу. Примеры манипуляций: обработка факторами роста, факторами дифференцировки, наращивание, генетическая модификация. Все эти процессы требуют для своего проведения строго контролируемых условий, по четко составленному, выверенному и отработанному плану, чтобы минимизировать любые отклонения.
По этой причине производство клеток должно осуществляться в соответствии с правилами надлежащей производственной практики (GMP) в асептических условиях, не допускающих внешней контаминации. Соблюдение таких условий должно быть валидировано. Достижение и поддержание таких условий должно четко контролироваться. - Невозможность исчерпывающего установления характеристик. Клетки — это сложные биологические объекты, обладающие бесчисленным множеством свойств и характеристик, некоторые из которых до сих пор не познаны.
Невозможность всесторонне охарактеризовать продукт производства означает, что он фактически определяется условиями и параметрами производства (процесс есть продукт). Следовательно, любые вариации процесса производства могут приводить к получению клеток с ранее неизвестными характеристиками, в том числе токсичными или неэффективными. Невозможность аналитически охарактеризовать клетки вынуждает сильно полагаться на результаты доклинических и клинических исследований.
Вместе с тем важно определить и отслеживать наличие (отсутствие) характеристик, имеющих решающее значение для профиля безопасности и эффективности (критичные показатели качества — CQA). В случае клеточных препаратов таковые включают жизнеспособность клеток, число клеток на дозу, степень дифференцировки и т. д. Кроме того, в сегодняшних условиях возможно определение широкого спектра генотипических и фенотипических характеристик, например с помощью секвенирования [23] генома отдельных клеток (single-cell genome sequencing) и имммунофенотипирования или современных вариантов сортировки клеток (например, флуоресцентной сортировки клеток [24]). Наконец, важно убедиться, что подвергшиеся всем запланированным манипуляциям клетки обладают требуемыми биологическими свойствами, например способностью вырабатывать биологически активные вещества, взаимодействовать с микроокружением или дифференцироваться в нужный клеточный тип. - Важность создания благоприятного микроокружения для клеток. В естественных условиях клетки находятся в очень специфическом окружении: они, как правило, связаны с соединительнотканными структурами, предоставляющими им опору, и получают различные химические и тактильные сигналы.
Для обеспечения хорошего «самочувствия» клеток и достижения целей производства нередко требуется использовать каркасы, матрицы или подложки для обеспечения 3D-структуры и физического контактирования клеток друг с другом, также может требоваться обработка клеток определенными биологически активными веществами и подбор питательных сред [25]. - Трудоемкость производства в расчете на дозу. Из сказанного ясно, насколько может быть трудоемким процесс производства клеточного препарата. С учетом того, что сегодня лекарственные препараты на основе клеток используются главным образом в аутологичных условиях или условиях подобранного донорства (matched donation), то есть когда пациент является аутодонором или реципиент подбирается в индивидуальном порядке, весь процесс производства фактически работает ради получения препарата для одного пациента. Это в свою очередь ведет к дороговизне получаемого продукта.
- Необходимость близкого расположения к пациенту из-за короткого срока годности и чувствительности к условиям транспортировки. Очень короткий срок годности как исходного материала (клеток человека), так и получаемого клеточного лекарственного препарата, а также их очень высокая чувствительность к условиям транспортировки и хранения затрудняют создание централизованного производства и требуют максимально возможного приближения производственных мощностей к месту оказания медицинской помощи.
Некоторые клеточные препараты могут требовать соблюдения труднодостижимых условий. Например, лекарственный препарат Kymriah (тисагенлеклейцел) должен храниться при температуре −120 °C или менее, тогда как лекарственный препарат Provenge (сипулейцел-T) мог храниться всего 18 часов при температуре 2–8 °C, а Strimvelis вообще хранится только 6 часов при температуре 15–30 °C [25]. Примечательна судьба лекарственного препарата Provenge. В 2015 г. он был выведен с рынка из-за коммерческой невыгодности проекта в связи с очень дорогим производством и сложной цепью поставок. Компания Dendreon построила три крупных производственных комплекса в США, при этом стоимость расходных материалов составляла около 70% в структуре выручки при цене 93 тыс. долл. за курс лечения. Вкупе с переоцененным спросом это привело к коммерческому провалу проекта и выведению его с рынка [17].
Кроме того, децентрализация производства требует наличие высококвалифицированного персонала в достаточном количестве в том месте, где будет осуществляться деятельность, что также может накладывать ограничения на реализуемость проекта. Эти ограничения особенно значимы при попытке организации производства клеточных препаратов в больничных условиях, где создание GMP-окружения требует дополнительных крупных вложений.
Все вышеперечисленные технологические ограничения в совокупности с относительно скромными достижениями в области регенеративной медицины с использованием клеточных лекарственных препаратов (то есть препаратов, содержащих модифицированные клетки) сильно тормозят развитие клеточных технологий, хотя спрос на эти препараты очень высок и продолжает быть сильно неудовлетворенным. Вместе с решением вопросов эффективности и безопасности клеточных препаратов отрасли предстоит решить проблемы их серийного выпуска: желательно, чтобы можно было использовать стандартизированные банки аллогенных клеток для универсального применения у разных пациентов вне зависимости от иммунологической совместимости или с возможностью индивидуализации. Такие банки должны быть охарактеризованы и максимально свободны от микробных контаминантов, а условия хранения и транспортировки не должны накладывать сильных ограничений на цепи поставок.
Генные терапии
Воздействие генной терапии по определению направлено на генетический аппарат клеток человека с терапевтической или профилактической целями. Европейское и американское законодательства рассматривают генотерапевтические препараты в качестве биологических [2], [3]. Тем самым антисмысловые олигонуклеотиды (-ерсены) [26] и препараты для РНК-интерференции (-сираны), получаемые при помощи химического синтеза, к генотерапевтическим не относятся. Классификационные подходы, содержащиеся в европейском законодательстве, перешли в законодательство ЕАЭС и выражены в части IV (раздел 17) приложения №1 к Правилам регистрации и экспертизы лекарственных средств для медицинского применения в Евразийском экономическом союзе [4].
С точки зрения того, в каких именно условиях происходит генетическая модификация, генные терапии принято разделять на препараты для модификации in vivo и препараты для модификации ex vivo. Первые — это неклеточные препараты, включающие препараты «голой» ДНК, препараты на основе невирусных векторов (например липосом), препараты на основе вирусных векторов или вирусоподобных частиц и препараты генетически модифицированных микроорганизмов (в первую очередь, бактерий). Они вводятся в организм человека с целью генетической модификации генетического материала клеток-мишеней.
При модификации ex vivo у пациента отбирают клетки, генетически модифицируют их и возвращают в организм (самого пациента-донора или других реципиентов). Помимо особенностей, присущих всем генотерапевтическим препаратам, для лекарственных препаратов на основе генетически модифицированных клеток характерны все те особенности, которые рассмотрены в отношении клеточных препаратов.
Проблемы производства и контроля качества генотерапевтических препаратов лежат в нескольких плоскостях и вызваны следующими факторами [18], [27–29]:
- Природа действующего вещества генотерапевтического лекарственного препарата. Это последовательность рекомбинантной нуклеиновой кислоты, которая непосредственно или опосредованно (после генетической модификации ex vivo) вводится в организм человека в целях регулирования, репарации, замены, добавления или удаления генетической последовательности [2]. С ней неразрывно связаны:
- неполное понимание всех генетических и эпигенетических зависимостей, которые в итоге приводят к полигенным заболеваниям (включая болезни сердечно-сосудистой системы, сахарный диабет, онкологические, нейродегенеративные и инфекционные заболевания), занимающим первые места в структуре заболеваемости и смертности, не связанной с травмами и насильственной смертью;
- уровень знаний в области молекулярной биологии, генной инженерии и смежных дисциплин.
- Отсутствие высокоспецифичных технологий доставки действующего вещества в клетки-мишени. Это значит, что пока не существует методов, которые могли бы с полной избирательностью доставить терапевтическую генетическую последовательность только в те клетки, которые нуждаются в генетической модификации. Внемишеневая доставка чревата различными вариантами токсичности, включая запуск канцерогенеза [29]. В этой связи важным аспектом разработки и производства становится создание подходящей и избирательной технологии доставки, которая может основываться на физических методах (например электропорации или сонопорации), химических методах (обработке поверхностно-активным веществами), носителях (липосомах) и биологических векторах (вирусах и вирусоподобных частицах).
При этом использование таких технологий доставки само по себе поднимает вопрос обеспечения их безопасности. Первые провалы применения генных терапий были связаны именно с иммуногенностью аденовирусных векторов-носителей: история Джесси Гелсингера [1] еще долго будет нависать над разработчиками этих препаратов.
Несмотря на сегодняшние достижения, всё равно не удается добиться ни полной специфичности в отношении клеток-мишеней, ни эффективной генетической модификации всех пораженных клеток. Недостаточная эффективность доставки терапевтического трансгена в целевые клетки особенно характерна для «защищенных» органов, таких как головной мозг.
Эти ограничения также требуют проведения комплекса аналитических испытаний для оценки того, в какие ткани и органы была доставлена генетическая конструкция, какова ее персистенция в организме, произошла ли интеграция в геном, каковы число копий генетической конструкции и ее идентичность, происходит ли экспрессия кодируемого генной конструкцией продукта. Вместе с тем с учетом ограничений современных аналитических технологий (чувствительности, специфичности, правильности, прецизионности, предела обнаружения и т. д.) получаемые результаты всегда приблизительны и не позволяют в полной мере охарактеризовать все происходящие в организме процессы. - Необходимость использования вирусных векторов. Сегодня это наиболее эффективный метод доставки генетического материала. Основные опасения при использовании большинства известных типов вирусных векторов — спонтанное восстановление вирулентности (например, если используются аденовирусные, герпесвирусные или ретровирусные векторы), потенциал беспорядочной интеграции (ретровирусов), а также недостаточная очистка от различных вирусов-контаминантов (например, от аденовирусов в случае аденоассоциированных вирусов). Кроме того, использование вирусных векторов чревато острым иммунным ответом — цитокиновым штормом [1].
Вследствие иммуногенности к вирусным векторам возможность их повторного использования ограничена, поэтому большинство генотерапевтических препаратов предназначено для однократного введения [27]. Если требуется неоднократное введение, необходимо продумать создание альтернативных систем доставки, что, по сути, предполагает создание нового лекарственного препарата с необходимостью полной характеристики его безопасности, эффективности, контроля производства и контроля качества.
В остальном получение и контроль качества генотерапевтических препаратов очень похожи на получение рекомбинантных белков [27]. Процесс получения предусматривает использование одного или нескольких процессов культивирования. Если используются невирусные методы доставки, то достаточно произвести только последовательность ДНК, как правило, с помощью E. coli. В случае же вирусных векторов необходимо также произвести вирусоподобные частицы или полноценные вирусные векторы. Это достигается использованием так называемых пакующих клеточных линий. Если вирусный вектор нуждается в помощнике, то может потребоваться производство вируса-помощника на клеточной линии, предназначенной для этого.
Если генотерапевтическим лекарственным препаратом являются генетически модифицированные (ГМ) клетки (например, CAR-T, ГМ-макрофаги [30], ГМ-NK-клетки [31]), то учитываются как особенности клеточных препаратов, так и бесклеточных генотерапевтических.
Ввиду сложности процессов производства, очень ограниченных возможностей по очистке от всевозможных примесей (в том числе, к примеру, не подвергшихся генетической модификации клеток, клеток других типов, присутствующих в образце) и инактивации микробных контаминантов, единственным действенным инструментом контроля качества препарата является правильно спланированный, всесторонне охарактеризованный процесс, который подвергся полноценной валидации. Валидация процесса производства предполагает подтверждение того, что итогом процесса производства станет лекарственный препарат требуемого качества. Для этого процесс повторяют много раз и всесторонне устанавливают характеристики входных материалов, промежуточных продуктов производства и конечного препарата. В итоге нужно подтвердить, что выбранные параметры процесса производства в должной мере отвечают цели, сформулированной во время разработки препарата [27].
Пример производства генной терапии
Разберем на примере генотерапевтического препарата Luxturna (воретигена непарвовек), зарегистрированного FDA в 2017 году для лечения одного из видов наследственной дистрофии сетчатки, как устроен процесс производства препарата и какие требования предъявляет к нему FDA. О самом препарате можно прочесть в предыдущей статье нашего цикла
Для получения лекарственного вещества — рекомбинантного аденоассоциированного вируса, содержащего терапевтический ген, — используются клетки почки человеческого эмбриона HEK293. Клетки клеточной линии HEK293 осуществляют биосинтез как терапевтического гена, так и компонентов системы доставки — вирусного вектора. Для этого их трансдуцируют тремя плазмидами, содержащими:
- экспрессионную кассету белка RPE65 (дефектного у пациентов);
- последовательности, кодирующие белки капсида вирусного вектора AAV2;
- последовательности вируса-помощника, упаковывающие кассету RPE65 в рекомбинантные частицы AAV2 (рис. 3).
Такой подход позволяет избежать использования живого вируса-помощника в процессе производства, что очень важно, ведь обычные методы удаления и инактивации вирусов невозможно применить к продукту на основе вирусного вектора.
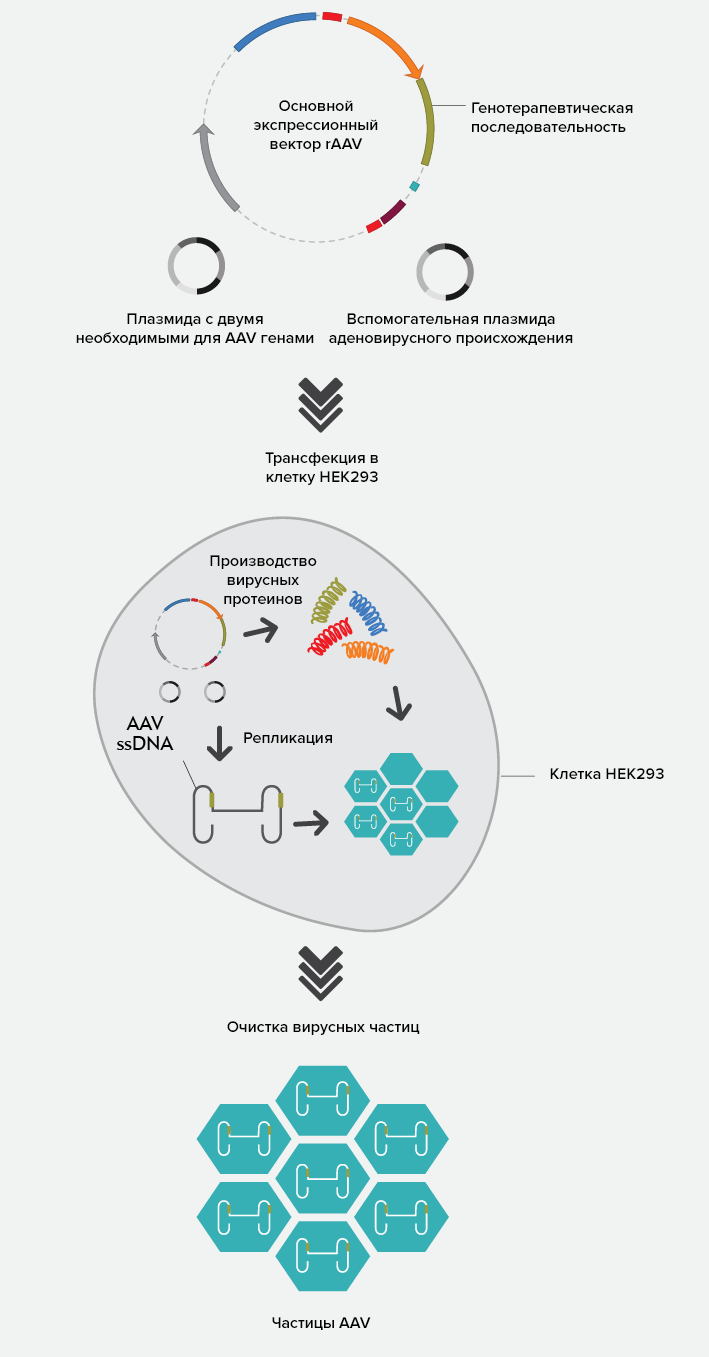
После культивирования в реакторе клетки собирают, подвергают фильтрации и катионообменной хроматографии. Затем препарат центрифугируют в градиенте плотности и еще раз фильтруют.
В публичном экспертном отчете FDA подробно описаны шаги производственного процесса, исходные материалы (банк клеток, плазмиды), приведены их спецификации и методы контроля. Характерно, что и до, и после подачи заявления на одобрение для продажи (в США она в случае биологических продуктов называется заявлением на лицензирование биопрепарата — biologics license application, BLA) FDA принимала очень активное участие в процессе, запрашивая у компании Spark множество уточнений и заставляя доделывать недостающие аспекты производства. Так, оказалось, что у компании были проблемы с обеспечением постоянства содержания одного из вспомогательных веществ в финальном продукте, однако после замечания FDA проблема была устранена.
Для всех стадий процесса были идентифицированы критичные параметры, критичные методы внутрипроизводственного контроля и диапазоны приемлемых значений. Затем была проведена валидация производственного процесса на одной серии продукта, произведенной в промышленном масштабе.
Продукт для клинических исследований производился на базе Филадельфийского детского госпиталя, а для коммерческого производства — на мощностях частной фирмы. Компания Spark провела исследования сопоставимости продуктов, полученных с помощью разных процессов производства, для доказательства того, что смена места производства не сказалась негативным образом на характеристиках безопасности и эффективности.
Директор компании в интервью рассказал, что досье для подачи BLA заняло 60 000 страниц, причем бóльшая часть объема была посвящена именно производству и методам его контроля. Компания разработала 41 испытание для оценки конечного продукта. Через два года после одобрения компания смогла перейти на биореакторы объемом 400 литров: это было непросто, так как нужно было научиться выращивать клетки в объеме, а не только в пристеночном слое. Теперь открылись возможности для масштабного производства генной терапии, что необходимо для более распространенных заболеваний.
Проблемы регулирования
Несмотря на то что идеи использования клеточных и генных препаратов в терапевтических целях не новы и насчитывают несколько десятилетий, более-менее систематическое их применение началось относительно недавно, и им еще только предстоит войти в основной арсенал используемых медицинских технологий.
Регуляторные требования, которые должны приниматься во время разработки, производства и контроля качества генотерапевтических и клеточных препаратов, учитывают различные особенности передовых терапий, риски и тяжесть их последствий. В частности, помимо упомянутого выше многоуровневого контроля микробиологических рисков, в отношении генотерапевтических препаратов действует отдельная отрасль законодательства в области генетически модифицированных организмов. Для оценки его соблюдения в Евросоюзе, к примеру, сформирована и действует отдельная система регулирования, отличная от системы регулирования лекарств [32–35]. Удовлетворение этих требований дополнительно осложняет разработку и производство генотерапевтических препаратов [36].
Беспрецедентными являются и меры контроля за препаратами передовой терапии после их введения пациенту. В ЕС наблюдение за такими пациентами должно длиться по меньшей мере 30 лет. Кроме того, должна быть обеспечена полная прослеживаемость от исходных материалов (например донорских клеток), до индивидуальных доз и далее до конкретных пациентов, и в обратном направлении, чтобы можно было однозначно идентифицировать «виновные» дозы и исходные материалы, если у пациента что-то пойдет не так, даже, к примеру, 25 лет спустя.
Регуляторные стандарты безопасности и эффективности препаратов передовой терапии, часть из которых воплощается в требованиях к производству и контролю качества, а также нередко индивидуальный характер разработок, делает их очень дорогими. Тем самым исследователи находятся под постоянным финансовым прессом из-за ограниченности ресурсов. С другой стороны, это вынуждает искать более экономичные методы разработки и миниатюризации производства, а также максимального приближения производственных мощностей к пациенту с целью экономии на транспортировке и хранении.
С экономическими аспектами тесно связана возможность воспроизведения препаратов для передовой терапии, которую целесообразно рассматривать с разных позиций, в том числе с точки зрения технической реализуемости, регуляторной реализуемости, экономической целесообразности, интеллектуальных прав и т. д. В целом движущая сила разработки воспроизведенных препаратов — возможность ценовой конкуренции с инновационным препаратом за счет экономии на разработке. Экономия на разработке исходит из ненужности повторения обширных доклинических и клинических исследований, которые были проведены разработчиком оригинального препарата, при условии того, что материальный носитель лекарственного препарата воспроизведен во всех критичных аспектах.
На сегодняшний день проблема воспроизведения клеточных препаратов и сложных генотерапевтических препаратов на основе вирусных векторов — задача труднореализуемая. Она требует приобретения образцов оригинального препарата, обратную инженерию его состава и структуры и дальнейшие масштабные лабораторные испытания для оценки успешности воспроизведения, прежде чем прототип будет готов к доклиническим и клиническим исследованиям. Уже на этом этапе разработчик столкнется со сложностями, поскольку образцы оригинального препарата могут быть запредельно дорогими, а в ряде случаев изготавливаться по индивидуальному заказу. Что касается трудностей воспроизведения таких сложных материальных объектов как вирусные векторы или живые клетки человека, следует отметить, что на сегодняшний день не воспроизводятся даже «простые» ферменты или факторы свертывания крови, которые, будучи сложнее моноклональных антител, тем не менее, гораздо проще по строению препаратов передовой терапии.
К техническим сложностям, которые сами по себе решаемы в долгосрочной перспективе, добавляются сложности научно-регуляторного характера. Нынешние препараты передовой терапии, как правило, предназначены для лечения редких заболеваний. Чтобы статистически подтвердить эквивалентность воспроизводимого препарата инновационному, в случае биопрепаратов, как правило, требуется выполнить сравнительные клинические исследования. Выборка для подтверждения эквивалентной эффективности может быть очень большой, достигая сотен человек, что для орфанных заболеваний может быть достаточно проблематично. Кроме того, обеспечение пациентов оригинальным препаратом тоже внесет большую лепту в стоимость исследования. В этом ключе гораздо дешевле и быстрее может оказаться разработка аналога с некоторыми второстепенными отличиями от оригинала, нежели полное воспроизведение [37–39]. Например, клиническая программа разработки аналогов рекомбинантных ферментов для лечения орфанных заболеваний или факторов свертывания крови обычно достаточно сжатая, требует всего несколько десятков пациентов и может даже не требовать сопутствующего сравнения с оригинальным препаратом. В этой связи гораздо важнее может быть воспроизведение терапевтического результата, а не самого лекарственного препарата.
Проблемы регулирования и производства генных и клеточных продуктов в России
Проблемы регулирования и производства генных и клеточных продуктов в России (рис. 4) обусловлены как общим отставанием в развитии фармацевтической отрасли и подходов к ее регулированию от ведущих зарубежных юрисдикций, так и специфичными проблемами, присущими именно этой области разработки, производства, изучения и регулирования терапевтических продуктов. Здесь мы остановимся только на специфичных проблемах.
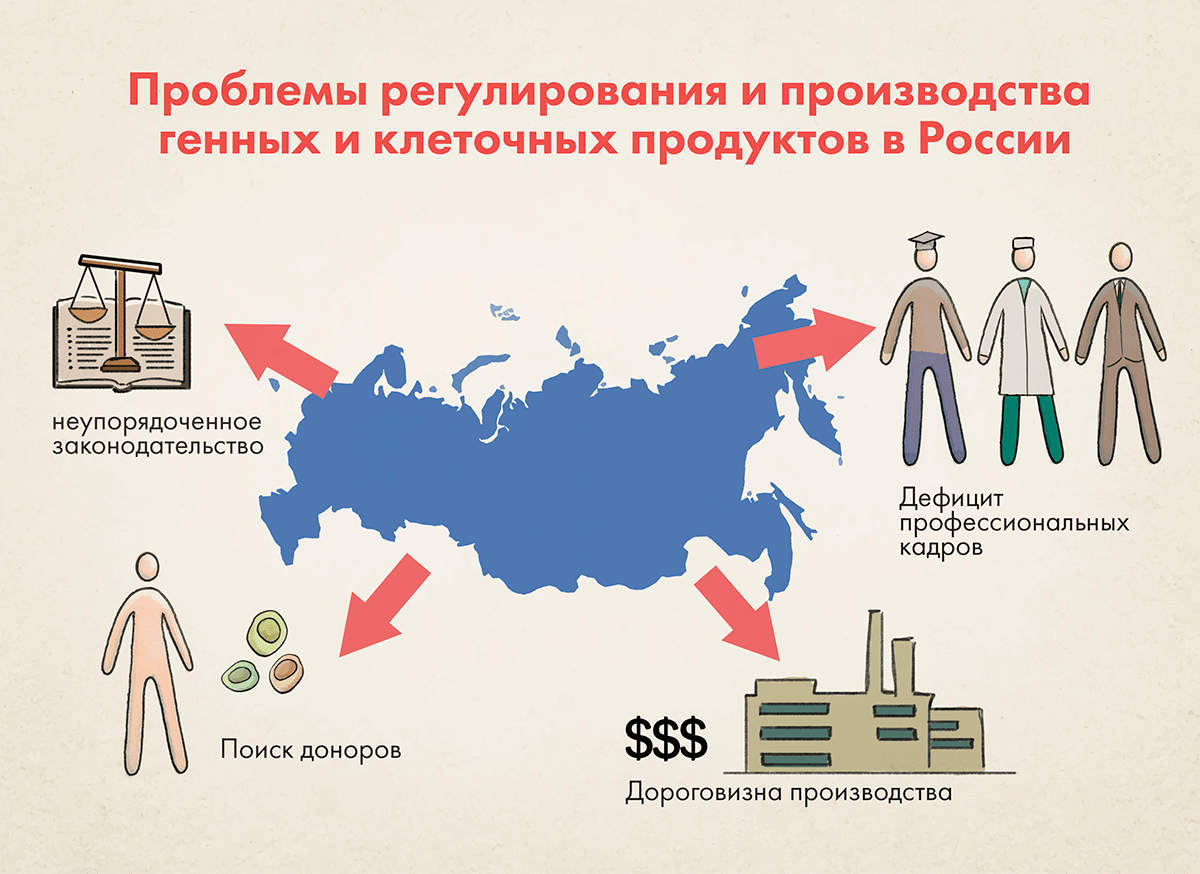
Основная проблема, трудно устранимая в кратко- и среднесрочной перспективе, — дефицит кадров. Эта сфера остро нуждается в биотехнологических и биохимических инженерах, специалистах в области биомедицинского анализа и визуализации, генетиках, фармацевтических микробиологах, специалистах в областях системной и вычислительной биологии, механики жидкостей, микрофлюидики и вычислительной динамики жидкостей. В нашей стране таких специалистов очень мало, а вузовские программы сильно отстают от ведущих мировых программ.
Следующим ограничением является дороговизна создания и поддержания производственных мощностей для производства клеточных препаратов, в которых должны соблюдаться строжайшие требования к асептическому производству, постоянству окружающих условий, разделению потоков производимых препаратов, специальным методам очистки и т. д. В случае индивидуализированных препаратов, например таких, как CAR-T, порой, весь производственный цикл осуществляется ради получения одной дозы для одного пациента. Точно так же выполняемый во время производства и по его завершении комплекс аналитических испытаний для оценки качества препарата на различных стадиях его получения реализуется для одной дозы или относительно небольшого их числа.
Не решены проблемы в области донорства и заготовки клеток, оценки и минимизации рисков при использовании генетически модифицированных организмов. Для ряда разрабатываемых препаратов в стране отсутствуют высокотехнологичные лаборатории, оснащение которых позволяло бы проводить широкий спектр испытаний на рецепторах, отдельных клетках, тканях и крупных животных (например, собаках, минипигах, нечеловекообразных приматах и др.), которые бы отвечали международным требованиям по обеспечению достоверности научных данных и гуманному обращению с животными.
Наконец, локальное законодательство в области генных и клеточных препаратов неупорядоченное и противоречивое. Отсутствует единое восприятие и отношение к ним как к подгруппе лекарственных препаратов, требующих дополнительных методов обеспечения безопасности, эффективности и качества. Основная проблема сегодняшней регуляторики этой сферы — отсутствие гармонизации с зарубежными подходами, требованиями и даже определениями. В частности, не выработаны частные критерии, предъявляемые к условиям и параметрам производства, оцениваемым показателям качества, целям, разновидностям и объему доклинических и клинических исследований в зависимости от вида разрабатываемого препарата, его терапевтического назначения и рисков для пациентов. Отсутствие специальных критериев для препаратов генной и клеточной терапий усложняет проведение исследований в полном объеме. Вместе с тем регуляторные проблемы — наименее сложные (их можно решить несколькими приказами Минздрава, тогда как, например, проблему недостатка кадров решить гораздо сложнее), поскольку сейчас ничто не мешает разработчикам ориентироваться на зарубежные требования, которые, пусть и обширны, но упорядочены и помогают сориентироваться в сложном процессе изысканий, разработки и проведения испытаний и исследований.
Мнение эксперта
Интервью Павла Гершовича «Биомолекуле»
Павел Гершович — директор Департамента разработки генотерапевтических препаратов компании BIOCAD, кандидат биологических наук, и.о. директора Научно-образовательного центра технологии рекомбинантных белков СПХФУ.
В компании BIOCAD Павел курирует работу по созданию генной терапии для лечения гемофилии, спинальной мышечной атрофии и других тяжелых наследственных заболеваний.
«Биомолекула»: Какова, по вашему опыту, трудоемкость процесса производства клеточных или генотерапевтических препаратов в расчете на дозу в сравнении с биотехнологическими белками?
Павел Гершович: Трудоемкость производства генотерапевтических и клеточных препаратов сильно варьирует в зависимости от природы генетического вектора и расчетной дозы препарата для введения пациенту. В сравнении с рекомбинантными белками, в том числе антителами, в среднем трудоемкость процесса пока остается значительно выше. Увеличение количества стадий производства неминуемо влечет за собой дополнительные трудозатраты. Это обусловлено тем, что для производства генотерапевтических лекарственных препаратов, или БМКП (биомедицинских клеточных продуктов — термин из российского законодательства; прим. ред.), используются генетические векторы различной природы, являющиеся полупродуктами финального биотехнологического процесса.
«БМ»: Рассматриваете ли вы децентрализацию производства как один из подходов для удешевления производства за счет приближения лекарства к пациенту?
ПГ: Децентрализация производства актуальна далеко не для всех генно-клеточных видов терапии. Такой подход может сильно упростить производство и клиническое применение большинства типов аутологичной передовой терапии, таких как БМКП для регенеративной медицины и CAR-T-терапии онкологических заболеваний. В системе производства, где пациент является единственным донором исходного материала и реципиентом конечного продукта, масштаб производства не имеет решающего значения. Если биомедицинская технология работает «у постели больного», это существенно сокращает затраты на производство и выводит доступность терапии на новый уровень. К сожалению, в нашей стране законодательство пока не позволяет в полной мере реализовать такую концепцию, что практически лишает шансов на выздоровление многих тяжело больных пациентов.
«БМ»: Какие регуляторные изменения компания хотела бы видеть в России?
ПГ: Гармонизация регуляторных требований по отношению ко всем видам препаратов передовой терапии на текущий момент является основой задачей. Мы имеем возможность анализировать и использовать накопленный опыт зарубежных коллег, чтобы создать все условия для роста и развития этой инновационной области фармацевтики в нашей стране.
«БМ»: Насколько российские практики производства, контроля качества отличаются от зарубежных? Насколько «Биокад» опирается на зарубежный опыт?
ПГ: «Биокад» неукоснительно соблюдает все требования регулятора, действующие на территории России. При этом мы придерживаемся стандартов качества, принятых на так называемых высоко регулируемых рынках. Производство любых типов лекарственных препаратов — это не статический процесс, а постоянное движение к совершенствованию технологических решений и системы контроля качества продукта, за которым стоит ежедневный кропотливый труд сотен специалистов.
Материал предоставлен партнёром — Департаментом разработки генотерапевтических препаратов компании BIOCAD
Заключение
Генные и клеточные терапии вносят пока незначительный общий вклад в арсенал терапевтических средств, уступая как низкомолекулярным лекарствам, так и рекомбинантным белкам. Однако эта область фармакологии активно развивается, следуя за научным прогрессом как в дисциплинах, лежащих в ее основе, так и в области производства и индивидуализации этих продуктов. Зарубежные регуляторы активно поддерживают развивающуюся отрасль, обобщая и формализуя свои рекомендации в виде научных и регуляторных руководств, направленных на систематизацию стратегии разработки, но с учетом индивидуальных особенностей новых препаратов, а также активно предлагают научные консультации, чтобы помочь компаниям выбрать наиболее оправданные пути продвижения вперед. Также у FDA и EMA есть специальных статусы для генотерапевтических и клеточных продуктов — например RMAT (Regenerative Medicine Advanced Therapy) у FDA, которые позволяют ускорить разработку продуктов и облегчить взаимодействие с агентствами.
В России некоторое улучшение регуляторной ситуации в области разработки лекарств ожидается с переходом на законодательство Евразийского экономического союза, которое во многих аспектах повторяет законодательство Евросоюза. В частности, есть надежда, что клеточная терапия будет регламентироваться не по закону 180-ФЗ, как «биомедицинские клеточные продукты», а как лекарства.
В будущем с точки зрения производства и контроля качества события будут развиваться в двух в определенной мере противоположных направлениях. Будут прорабатываться методы крупномасштабного производства препаратов, предназначенных для лечения распространенных заболеваний. Параллельно будут развиваться подходы индивидуализированного производства для получения препаратов под нужды конкретных пациентов.
В следующей статье цикла мы расскажем подробнее о клеточных терапиях, в первую очередь, конечно, о Т-клетках с химерным антигенным рецептором (CAR-T).


