

Резюме | Окислительный стресс — это звено патогенеза таких заболеваний как атеросклероз, ХОБЛ, болезнь Альцгеймера, онкологии и многих других. И хотя ряд антиоксидантных молекул демонстрировали терапевтический потенциал в доклинических исследованиях, результаты их клинических испытаний оказались разоваровывающими. Только глубокое понимание механизмов действия антиоксидантов и ситуаций, когда они эффективны, может обеспечить их рациональное использование и, как следствие, фармакологический успех. В данном обзоре авторы рассматривают связь окислительного стресса и редокс-сигналинга с патологией, а также механизмы, по которым окислительный стресс приводит к заболеваниям, и работу систем антиоксидантной защиты, ограничители ее эффективности и способы ее повышения с помощью пищевых добавок и потенциальных лекарств.
Введение
Гельмут Сис впервые определил “окислительный стресс” как дисбаланс между синтезом окислителей и возможностями антиоксидантной защиты, который приводит к повреждению биологических систем [1]. С тех пор область редокс-биологии (биохимии редокс-пар, прим перев.) развилась от концепции окислительного стресса при патологии до редокс-сигналинга в нормальной физиологии [2-4].
Доказано участие окислительного стресса в развитии широкого спектра заболеваний, таких как атеросклероз, хроническая обструктивная болезнь легких (ХОБЛ), болезнь Альцгеймера и онкологические заболевания. Это позволило выявить многочисленные механизмы повреждения клеток оксидантами [5]. Однако степень участия окислительного стресса в патогенезе различных заболеваний сильно варьирует, поэтому польза от повышения антиоксидантной защиты при некоторых заболеваниях может быть ограничена.
Окислительный стресс включает в себя реакции с участием так называемых активных форм кислорода и азота (вставка 1). Понимание того, кто из них повреждает макромолекулы, помогает развить терапевтические подходы в антиоксидантной защите. Однако до сих пор использование малых молекул в терапевтических целях не приносит успеха, в основном из-за чрезмерно оптимистичных и неверных предположений о том, как работают антиоксиданты [6]. Например, инактивация гидроксильного радикала (•OH) оказалась неэффективной, но предотвращение его образования путем торможения синтеза перекиси водорода (H2O2) может обеспечить необходимую защиту от повреждений. Одно из основных заблуждений в области окислительного стресса касается инактивации супероксида (O2•-) или H2O2 малыми молекулами, которые также неэффективны внутри клеток. Это связано с тем, что собственные антиоксидантные ферменты организма дают лучшую защиту и реагируют с этими оксидантами в тысячи и миллионы раз быстрее, чем малые молекулы [6,7]. Однако во внеклеточных жидкостях, где антиоксидантных ферментов нет, инактивация O2•- и H2O2 (но не •OH) возможна с помощью миметиков супероксиддисмутазы (СОД) и каталазы, о чем подробнее будет сказано далее.
Важно разобраться в ограничениях, которые привели к неудачам в клинических испытаниях и в том, как антиоксиданты могут стать эффективнее, если знать, где, когда и в какой степени окислительный стресс вовлечен в патогенез заболевания. По большей части антиоксидантная защита внутри клеток обеспечивается не экзогенными или эндогенными малыми молекулами, а антиоксидантными ферментами, использующими свои специфические субстраты для восстановления оксидантов. Таким образом, основные терапевтические возможности заключаются в предотвращении синтеза оксидантов, вызывающих серьезное повреждение макромолекул, ингибировании их последующих сигналов, запускающих воспаление или гибель клеток, а также в увеличении количества антиоксидантных ферментов и их субстратов. В настоящее время проводятся клинические испытания эбселена, имитатора глутатионпероксидазы (ГП) для лечения болезни Меньера в фазе II (NCT02603081); GC4419, миметика СОД для лечения плоскоклеточного рака в фазе I (NCT01921426); и сульфорафана, активатора фактора транскрипции NRF2, для лечения ХОБЛ в фазе II (NCT01335971) и др.
В данной статье рассматриваются взаимосвязи между окислительным стрессом, редокс-сигналингом и патологиями, а также представлен обзор механизмов, с помощью которых окислительный стресс может способствовать развитию заболеваний. Авторы раскрывают современное понимание механизмов антиоксидантной защиты, описывают факторы, ограничивающие их эффективность, а также освещают новые терапевтические подходы для воздействия на эти процессы. Глубокое понимание работы оксидантов с учетом ограничений и возможностей антиоксидантной терапии приведет к повышению качества терапевтических вмешательств.
Авторы данного обзора подразумевают под окислительным стрессом ситуацию, в которой оксиданты неферментативно повреждают макромолекулы, включая белки, нуклеиновые кислоты и липиды клеточных мембран. Данный обзор сосредоточен только на факторах, которые либо предотвращают выработку оксидантов, либо обеспечивают их эффективное удаление. Основными мишенями являются O2•-, H2O2 и гидроперекиси липидов. Нейтрализация этих мишеней предотвращает производство более реактивных •OH, пероксинитрита (ONOO—) и гипогалогеновых кислот (HOX). Хотя синтез ONOO- можно ограничить путем ингибирования производства оксида азота (•NO), сам •NO слишком важен для поддержания нормальной физиологии, и лучшим подходом является ограничение чрезмерного производства O2•-.ГлоссарийОкислительный стресс — дисбаланс между синтезом оксидантов и способностью организма предотвращать окислительное повреждение.
Редокс-сигналинг — процессы передачи сигналов, в которых в качестве вторичных мессенджеров выступают оксиданты.
Антиоксидантная защита — предотвращение или устранение окислительного повреждения.
Антиоксидантные ферменты — строго говоря, это ферменты, которые инактивируют оксиданты. В широком смысле (подразумевается в данном обзоре) — ферменты, которые препятствуют окислительному повреждению или же устраняют его последствия.
Транскрипционный фактор NRF2 — Е2-родственный ядерный фактор 2, управляющий активацией большого числа антиоксидантных ферментов в покое и во время окислительного стресса.
Антиоксидантная терапия — лечение при помощи соединений, усиливающих антиоксидантные защиты организма.
Пневмонит — воспаление легких, вызванное раздражением ткани легких, заболеванием, инфекцией, лучевой терапией или аллергией.
Ишемия-реперфузия — остановка кровоснабжения и затем его возобновление.
Аутофагия — механизм, при помощи которого утилизируются ненужные или поврежденные компоненты клетки.
Роль окислительного стресса в заболеваниях
Есть два ключевых механизма, посредством которых окислительный стресс способствует развитию заболеваний. Первый включает синтез АФК — а именно •OH, ONOO— и HOCl—, которые прямо окисляют макромолекулы (липиды мембран, структурные белки, ферменты и нуклеиновые кислоты), приводя к нарушению функционирования клеток и их смерти. Вторым механизмом является редокс-сигналинг (Вставка 2). Оксиданты, в частности H2O2, синтезируются клетками под влиянием физиологических стимулов и могут действовать как вторичные мессенджеры [8]. Но при окислительном стрессе, когда синтез H2O2 выходит за физиологические рамки, редокс-сигналинг приобретает патологический характер [4].
Оба этих механизма окислительного стресса могут происходить в рамках одного заболевания, например, диабета, когда накопление конечных продуктов гликирования и аберрантная (искаженная, патологическая — прим. перев.) активация сигнальных путей клеточного стресса приводят к осложнениям [9]. Кроме этого, при окислительном стрессе O2•- (источник 10) и ONOO— (источник 11) высвобождают ионы железа из белков, что вместе с избыточным синтезом H2O2 увеличивает количество продуктов окисления липидов, включая 4-гидрокси-2-ноненаль (HNE), который также может провоцировать аберрантный сигналинг [12].
Окислительный стресс ассоциируется с широким диапазоном заболеваний, которые были разделены на две категории: те, где окислительный стресс является первичной причиной (включая атеросклероз и повреждения, вызванные радиацией и паракватом); и те, где окислительный стресс вносит вторичный вклад в прогрессирование заболевания (ХОБЛ, артериальная гипертензия и болезнь Альцгеймера). Однако роль окислительного стресса во многих заболеваниях изучена не до конца, и потому данное разделение не окончательное.Вставка 1 | Механизмы окислительного стрессаОкислительный стресс могут вызывать эндогенные и экзогенные соединения [276]. Под термином активные формами кислорода (АФК) объединяют молекулы, получаемые из O2: супероксид (O2•−), перекись водорода (H2O2), гидроксид радикал (•OH), озон и синглетный кислород. Использование термина АФК, как если бы он подразумевал одно химическое соединение, приводит ко многим неточным заключениям, так как химические свойства этих молекул существенно различаются.
Синтез O2•− путем одноэлектронного восстановления O2 происходит благодаря утечке электронов из цепи переноса электронов митохондрий, особенно из реакции убисемихинона (QH•−) [277] (реакция 1):
QH∙− + O2 ↔ Q + O2∙−
и НАДН-оксидазы, которая катализирует реакцию 2 (источники 232, 278):
NADPH + 2O2→NADP+ + H+ + 2O2∙−
НАДН-оксидазы (NOX4, DuOX1 и DuOX2), а также некоторые другие флавопротеиновые ферменты, восстанавливают O2 до H2O2, поставляя второй электрон для O2•− до того, как он успеет покинуть их активный центр [279].
Главным источником H2O2 является дисмутация O2•−. Это быстрая реакция с константой около 105 M−1 s−1, которая катализируется супероксид-дисмутазами до 2 × 109 M−1 s−1 (реакция 3):
2O2∙− + 2H+ → H2O2 + O2
Скорость синтеза H2O2 в большой степени определяет, будет происходить редокс-сигналинг, окислительный стресс или не произойдет значимого окисления чего-либо. В восстановлении H2O2 участвуют 15 ферментов, включая каталазу (реакция 4):
2H2O2 → 2H2O + O2,
пять пероксиредоксинов, использующих тиоредоксин (небольшой белок с двумя цистеинами, Trx(SH)2) или восемь глутатионпероксидаз и пероксиредоксин 6, которые используют глутатион (трипептид, γ-глутамил-цистеинил-глицин, GSH) (реакции 5 и 6):
H2O2 + 2Trx(SH)2 → TrxS2 + 2H2O
H2O2 + 2GSH → GSSG + 2H2O
где TrxS2 — тиоредоксин дисульфид и GSSG — глутатион дисульфид.
H2O2 не может легко окислять большинство молекул, но она быстро реагирует с металлами переходной валентности, такими, как железо, с образованием гидроксил радикала (реакция 7, часто называемая реакцией Фентони) [280]:
H2O2 + Fe2+ → OH− + Fe3+ + ∙OH
Гидроксил радикал является чрезвычайно сильным окислителем, который может быстро окислять любую молекулу, до которой доберется.
Также ответственна за окислительный стресс цепная реакция перекисного окисления липидов, которая запускается ∙OH (реакции 8–10):
LH + ∙OH → L∙ + H2O
L∙ + O2 → LOO∙
LOO∙ + LH → LOOH + L∙
где LH — это липид с аллильными водородами, которые присутствуют в полиненасыщенных жирных кислотах, в том числе арахидоновой.
Супероксид может высвобождать железо из железо-серных белков, которое запускает реакцию 7. Однако главный механизм, через который относительно слабый оксидант O2•− способствует развитию окислительного стресса — это синтез из него H2O2 и пероксинитрита (ONOO−), который показан в реакции 11:
O2∙− + ∙NO → ONOO−
где ∙NO — оксид азота (II).
Опасность окислительного стресса исходит не прямо от свободных радикалов ∙NO и O2∙−, а от не-радикала — протонированной формы пероксинитрита — пероксиазотной кислоты (ONOOH). Пероксиазотная кислота является сильным окислителем, ее реактивность опосредована промежуточными соединениям, появляющимися в процессе ее распада диоксида азота ∙NO2 и ∙OH. (реакция 12):
HNOO− ↔ + ∙NO2 + ∙OH → NO3− + H+
∙NO2 может отщеплять водород, как это делает ∙OH или добавлять его к некоторым молекулам, например, тирозинам белков с образованием нитротирозинов, нарушающих их функцию. ONOO− также может высвобождать железо из белков с железо-серными кластерами [11], способствуя синтезу ∙OH из H2O2 (реакция 7). ∙NO2 и ∙OH окисляют все молекулы без разбора, что и создает хаос, называемый окислительным стрессом. Ввиду большой скорости их реакций, лучший подход здесь — это предотвращение образования ∙NO2 и ∙OH.
Последние оксиданты, которые мы рассмотрим, это гипогалогеновые кислоты (HOX), которые образуются из H2O2 в реакции 13, катализируемой миелопероксидазами фагоцитов:
H2O2 + X− + H+ → HOX + H2O
где X — может быть Cl−, Br− или даже SCN− (источник 281).
Они играют главную роль при повреждении тканей с фагоцит-опосредованным воспалением.
Окислительный стресс как первичная причина патологий
Окислительный стресс может быть первичной причиной повреждения и заболевания. Однако важно отметить, что с момента начала окислительного повреждения антиоксидантная терапия часто уже не может затормозить разрушение тканей, поскольку в развитии патологии начинают лидировать другие факторы.
Радиационное поражение легких. Ранний пневмонит с последующим фиброзом — частый побочный эффект лучевой терапии рака легких и пищевода [13]. Под воздействием радиации в клетках происходит гомолитическое расщепление молекул воды с высвобождением •OH, который затем окисляет макромолекулы и запускает воспалительный ответ, в результате чего клетки воспаления инфильтрируют ткань (пневмонит) и паренхима легкого гибнет. Со временем такой патологический редокс-сигналинг и хронический синтез цитокинов приводят к накоплению коллагена и фиброзу легких [14]. Кроме того, на модели лучевого поражения легких у крыс спустя месяцы после воздействия наблюдалось повышенное окисление липидов и ДНК (8-гидрокси-2’-деоксигуанозин).
Отравление паракватом. Окислительный стресс также является основной причиной токсичности широко распространенного гербицида параквата. При попадании внутрь паракват активно захватывается альвеоцитами 2 типа с развитием пневмонита и прогрессирующего фиброза легких с неблагоприятным прогнозом. Паракват также повреждает печень, почки и другие органы. Долгосрочное воздействие параквата на организм связано с развитием болезни Паркинсона [16]. Токсичность параквата возникает в результате циклических реакций окисления-восстановления, генерирующих O2•- (источник 17).
Атеросклероз. При атеросклерозе в интиме артерий формируются бляшки, что со временем сужает просвет артерий с возникновением инсульта и инфаркта миокарда. Собрано достаточно доказательств решающей роли окислительного стресса в патогенезе атеросклероза. С момента первого обнаружения гидропероксидов липидов в пораженной атеросклерозом аорте человека [18] множество исследований показали повышение уровня окисленных липидов и других маркеров окислительного стресса в области атеросклеротических поражений сосудов. Например, есть данные, что 20% линолеата холестерина (18:2) в свежих изолятах атеросклеротических бляшек человека были окисленными, чего не наблюдалось в непораженных артериях [19]. Кроме того, было обнаружено, что у 50% пациентов с атеросклерозом был повышен уровень HNE-модицифированных липопротеинов низкой плотности (ЛНП) по сравнению со здоровыми добровольцами [20]. Более того, продукты пероксидации арахидоновой кислоты — изопростаны — были как минимум в 5 раз повышены в областях атеросклеротического поражения артерий по сравнению с пупочной веной, а окисленная линоленовая кислота обнаруживалась только в местах поражений [21]. Окислительный стресс отвечает за превращение холестерола ЛНП в атерогенную форму окисленных ЛНП, которые играют решающую роль в инициации и поддержании воспалительной реакции и рекрутировании лимфоцитов в область поражения и способствует развитию атеросклероза через активацию гладких миоцитов и низкую доступность •NO [22].
Окислительный стресс играет вторичную роль в прогрессировании заболеваний
При многих заболеваниях окислительный стресс развивается как следствие других факторов, инициирующих патологию. К примеру, окислительный стресс запускается ксантиноксидазой при ишемии-реперфузии и избыточной продукцией O2•- или H2O2 NADPH-оксидазами (NOX) в рамках воспалительного ответа при повреждении тканей.
Окислительный стресс может нарушать работу сигнальных путей и влиять на многие биологические процессы через модификацию белков, усиление воспаления, запуск апоптоза и аутофагии, нарушение функционирования митохондрий и так далее. Эти эффекты часто ускоряют прогрессирование патологических процессов и усугубляют симптомы заболеваний, что далее будет разобрано на наглядных примерах.
Хроническая обструктивная болезнь легких. ХОБЛ подразумевает прогрессирующий и необратимый хронический бронхит и/или эмфизему. Курение сигарет, как источник большого числа окислителей, является главной причиной ХОБЛ. В основе патогенеза ХОБЛ лежит подавление окислительным стрессом а1-антитрипсина, что снижает его способность ингибировать эластазу нейтрофилов [23]. Кроме того, хроническое воздействие оксидантов сигаретного дыма вызывает воспаление и другие патологические каскады, такие как клеточная смерть и фиброз [14]. Источники оксидантов могут быть как экзогенными (например курение или загрязнение воздуха), так и эндогенными (например, NOX митохондрии, индуцируемая синтаза оксида азота (iNOS)и миелопероксидаза) [14].
Повышенные уровни оксидантов и продуктов ПОЛ, включая 8-изопростан, постоянно обнаруживаются в конденсатах выдоха пациентов, больных ХОБЛ [24]. Кроме того, уровень HNE (HNE аддуктов) был как минимум на 50% выше в дыхательных путях и альвеоцитах, эндотелиоцитах и нейтрофилах у пациентов с ХОБЛ по сравнению со здоровыми [25]; также у этих пациентов в моче было повышено содержание 8-гидроксидеоксигуанозина (8-OHdG) — маркера окисления ДНК [26]. Функция легких пациентов обратно коррелировала с интенсивностью окислительного стресса [25]. В совокупности эти данные подтверждают, что окислительный стресс у пациентов с ХОБЛ происходит как в легких, так и системно, и вовлечен в патогенез заболевания.
Идиопатический легочный фиброз. Патогенез идиопатического легочного фиброза (ИЛФ) заключается в диффузном прогрессирующем мезенхимальном фиброзе и вялотекущем воспалении неизвестной этиологии. Множество исследований показали присутствие окислительного стресса при ИЛФ. Маркеры окислительного стресса, такие как H2O2, 8-изопростан, 8-изопростагландин-F2α (8-iso-PGF2α) и этан в большом количестве обнаруживались в конденсате выдыхаемого воздухе пациентов с ИЛФ [27]. Кроме того, в бронхоальвеолярном лаваже пациентов с ИЛФ были повышены уровни 8-изопростана [28] и окисленных белков [29] в пять и два раза соответственно. Также при ИЛФ были значимо повышены уровни HNE в легких и 8-изопростана в крови [31]. Уровень глутатиона (GSH) в бронхоальвеолярной лаважной жидкости и мокроте пациентов с ИЛФ в был четыре раза ниже, чем у здоровых [32], что указывает на дефицит этого важного компонента антиоксидантной защиты при ИЛФ. Синтез H2O2, по-видимому, происходит в основном за счет NOX4 (источник 33) и дисфункциональных митохондрий, а синтез глутатиона снижается под действием сигнальных путей TGFβ [35]. Все больше данных свидетельствуют о том, что окислительный стресс играет важную роль в ИЛФ, способствуя фиброгенезу через апоптоз альвеоцитов, активацию миофибробластов и воспаление [36]. Помимо окислительного стресса, патогенез ИЛФ также включает апоптоз, сенесценцию, эпителиально-мезенхимальный переход, эндотелиально-мезенхимальный переход, миграцию эпителиальных клеток, повышенную продукцию хемокинов, цитокинов и факторов роста, а также дисфункцию митохондрий, стресс эндоплазматического ретикулума, гипоксию и воспаление [37]. Эти механизмы взаимосвязаны, и окислительный стресс является важным компонентом патогенеза ИЛФ.
Артериальная гипертензия. Гипертонической болезни способствуют множество факторов риска, таких как диета, курение, образ жизни, генетика и сопутствующие заболевания. При этом более 90% случаев — это эссенциальная гипертензия неясной этиологии. Однако на молекулярном уровне окислительный стресс является общей чертой этого состояния. Экспериментальные исследования показывают, что при гипертонии оксиданты в основном образуются при участии различных изоформ NOX [38]. В крови пациентов с гипертонией были значительно повышены маркеры окислительного стресса, включая H2O2 (источник 39), соотношение уровней дисульфида глутатиона (GSSG) и глутатиона, малоновый диальдегид (продукт перекисного окисления липидов) [40] и 8-изопростан [41]. H2O2 играет определенную роль в развитии и прогрессировании гипертонии, влияя на сигнальные пути ангиотензина II, NO и другие внутриклеточные процессы [42]. Однако, причинно-следственная связь между окислительным стрессом и гипертонией еще не установлена.
Сахарный диабет 2-го типа. У пациентов с СД2 окислительный стресс проявляется микро- и макроангиопатиями [43]. Обнаружено, что в плазме пациентов с СД2 значительно повышены маркеры окислительного стресса, включая соотношение OxLDL и LDL [44], 8-OHdG [45], 8-изо-PGF2α [46], белковые карбонилы [47] и конъюгаты глутатиона с гемоглобином [48], а также уровни 8-OHdG и 8-изо-PGF2α в моче [49]. Увеличение количества оксидантов при СД2 происходит из-за дисфункции митохондрий [50] и NOX1 (источник 51), которые являются следствием диабетических нарушений — гипергликемии и дислипидемии.
Болезнь Альцгеймера. Болезнь Альцгеймера характеризуется прогрессирующим внеклеточным накоплением бляшек β-амилоида и нейрофибриллярных клубков внутри нейронов. Было выявлено несколько факторов риска развития болезни Альцгеймера (возраст, генетика, пол, травмы и загрязнение воздуха), но точная причина остается неясной. При этом появляется все больше данных о том, что ключевую роль выполняет окислительный стресс [52].
Во многих исследованиях наблюдали повышенный уровень окислительного стресса в мозге пациентов с болезнью Альцгеймера, включая повышенные уровни F2-изопростана-α в спинномозговой жидкости [53] и лобных и височных долях [54], акролеина в миндалине и гиппокампе/парагиппокампальной извилине [55], и HNE в ликворе желудочков [56], гиппокампе и нижней теменной доле [57], и коре [58]. Также в лобных, теменных и височных долях мозга пациентов с болезнью Альцгеймера обнаружили усиленное окисление ядерной и митохондриальной ДНК относительно соответствующих по возрасту контрольных субъектов [59]. Кроме того, у пациентов с болезнью Альцгеймера значительно увеличено количество окисленных белков в гиппокампе [60] и карбонилов белков в коре головного мозга [58]. Утверждается, что повышенному синтезу оксидантов способствуют Aβ(1-42) [61], активация микроглии [62], накопление железа [63] и дисфункция митохондрий [64].
Рак. Оксиданты участвуют в различных фазах онкогенеза, включая трансформацию нормальных клеток в опухолевые, а также рост, пролиферацию, инвазию, ангиогенез и метастазирование опухолевых клеток. Это происходит из-за нарушения работы сигнальных путей, повреждения ДНК и усиления воспаления [65]. С другой стороны, окислительный стресс может сам запускать апоптоз и ферроптоз, защищая от трансформации и тем самым предотвращая онкогенез [65]. Одновременно окислительный стресс является основным повреждающим механизмом радиации (см. подраздел «Повреждение легких радиацией» выше) и многих химиотерапевтических препаратов [66].
Таким образом, окислительный стресс участвует почти во всех аспектах существования опухоли. Опухолевые клетки производят больше оксидантов, чем нормальные, и поэтому подвергаются повышенному действию окислительного стресса. Основными поставщиками избыточных количеств оксидантов в опухолевых клетках служат митохондрии [67], NOX4 (источник 68) и 5-липоксигеназа [69]. Оксиданты могут также поступать из нормальных клеток опухолевой массы или соседних, таких как эндотелиальные клетки и воспалительные иммунные клетки. Повышение уровня окислительных маркеров наблюдается при различных видах рака. Например, было показано, что в выдыхаемом воздухе пациентов с немелкоклеточным раком легких содержится больше H2O2, чем у здоровых [70]. Кроме того, повышенные уровни 8-OHdG [71] были обнаружены в тканях рака молочной железы по сравнению с аналогичными образцами здоровой ткани, а значительное повышение 8-OHdG наблюдалось при раке простаты [72] и раке легкого [73].Вставка 2 | Редокс-сигналинг, гомеостаз и антиоксидантные защитыСуть редокс-сигналинга состоит в специфическом взаимодействии сигнальных белков с H2O2 или другими электрофилами, которые в данном случае работают как вторичные мессенджеры. Экзогенные и эндогенные источники H2O2 и другие электрофилы могут быть частью окислительного стресса, однако чтобы редокс-сигналинг был скорее физиологическим, а не патологическим, необходима регуляция и специфичность. В отличие от окислительного стресса, редокс-сигналинг не приводит к повреждению. такое явление мы называем “пара-гормезис” [101], этот термин близок к “окислительному эустрессу” [3].
Поддержание редокс-гомеостаза очень важно для нормального функционирования клетки. Несмотря на название, гомеостаз не подразумевает, что все остается неизменным, хотя действительно, необходим баланс между окислителями и восстановителями (глутатион, тиоредоксин и НАДФН, субстратами антиоксидантных ферментов) [101]. Таким образом, заболевания с участием окислительного стресса можно рассматривать как нарушения редокс-гомеостаза, таким примером может быть сахарный диабет 2 типа [9]. По определению Kelvin Davies [282] адаптивный гомеостаз включает в себя рост антиоксидантной защиты, спровоцированный изменением редокс-гомеостаза и редокс-сигналинга. Так как окислительный стресс может стимулировать те же сигнальные пути, что и физиологический редокс-сигналинг, последний также может протекать и в рамках патологии. Разница заключается в том, что редокс-сигналинг в рамках окислительного стресса будет протекать неконтролируемо и сопровождаться неспецифическим повреждением.
Эффективность антиоксидантных систем в поддержании гомеостаза измеряется в способности поддерживать синтез и нейтрализацию супероксида (O2•−), H2O2 и оксида азота (•NO) в диапазонах концентраций, которые не позволяют образовываться значительным количествам пероксинитрита и гидроксил радикала [101]. Эта система несовершенна, о чем говорит небольшое накопление окисленных белков с возрастом. Независимо от этого, возможность индуцировать ферменты, удаляющие O2•−, H2O2 и поврежденные белки, то, что Davies называет “адаптивным гомеостазом”, представляет собой основное направление по усилению антиоксидантной защиты и будет далее рассмотрено в этом обзоре [282].
Синдром системного воспалительного ответа (ССВО). ССВО это избыточный воспалительный ответ всего организма на инфекционные патогены или неспецифические повреждения [74]. При ССВО высвобождается огромное количество оксидантов и провоспалительных цитокинов, что приводит к обратимой или же необратимой дисфункции органов, или даже к смерти. Сепсис, который изучен гораздо лучше, имеет общие черты воспаления и окислительного стресса с ССВО, вызванного неинфекционным повреждением, и может рассматриваться как ССВО, вызванный инфекцией.
Сообщается, что у пациентов с тяжелым сепсисом значительно повышался уровень F2-изопростанов [75], HNE [76] и 8-OHdG [77] в плазме. У больных ОРДС на фоне ССВО наблюдались повышенные уровни 8-изо-PGF2α в конденсате выдыхаемого воздуха [78] и нитротирозина в бронхопульмонарном лаваже [79]. При сепсисе в зависимости от тканей и\или клеток источниками оксидантов могут быть iNOS (также известная как NOS2) [80], NOX [81], ксантиноксидаза [82] и митохондрии с нарушенной функцией [83]. Кроме этого, при сепсисе снижается уровень таких антиоксидантов как витамин C [84], витамин E [85] и GSH [86].
Ишемия-реперфузия. Хотя своевременная реперфузия необходима для предотвращения последствий ишемии, она может вызывать обширные повреждения близлежащих тканей и отдаленных органов ввиду запуска системного воспалительного ответа. Ишемическо-реперфузионное повреждение (ИРП) играет ведущую роль в патофизиологических изменениях при таких критических состояния, как инфаркт миокарда, инсульт и трансплантация органов. Молекулярные механизмы в основе ИРП включают в себя воспалительный ответ и окислительный стресс. В фазу ишемии дефицит кислорода и нутриентов приводит к накоплению гипоксантина, высвобождению ионов кальция и провоспалительных цитокинов, активации ксантиноксидазы; в фазу реперфузии в системе гипоксантин/ксантиноксидаза [87], митохондриях, iNOS (NOS2) и NOX [88] эндотелиоцитах, нейтрофилах и локально в клетках [89] запускается продукция NO, ONOO—, O2•- и других оксидантов. Так, у пациентов с инфарктом миокарда после тромболизиса были повышены уровни маркеров окислительного стресса (включая 8-изо-PGF2α в моче) по сравнению со здоровыми индивидами того же возраста, пациентами со стабильной стенокардией [90] и пациентами с коронарной ангиопластикой после реперфузии сонной артерии [91]. Исследование 66 человек с инсультом и 132 человек в контрольной группе показало, что содержание F2-изопростанона в плазме и моче немедленно повышалось и сохранялось выше нормы вплоть до 7-го дня от начала ишемического инсульта [82]. Уровень 8-OHdG в моче также был повышен после реперфузии при остром инфаркте миокарда [83]. Следует отметить, что большинство измеренных окислительных маркеров были системными, и лишь в немногих исследованиях определялось их наличие в очаге поражения.
Системы антиоксидантной защиты и терапевтические подходы
В процессе эволюции живые организмы развили системы защиты от оксидантов — антиоксидантные ферменты, а также пути пополнения их субстратов и восстановления поврежденных структур. Эти системы индуцируются в ответ на окислительный стресс и другие электрофилы, и также растет способность клеток обезвреживать оксиданты и/или электрофилы и восстанавливаться после окислительного повреждения. Соединения, которые усиливают подобные системы самозащиты клеток, составляют основу антиоксидантной терапии.
Обширные исследования были сосредоточены на регуляторных механизмах антиоксидантных ферментов и их возможной роли при заболеваниях. Существует ряд редокс-чувствительных транскрипционных факторов, которые вовлечены в индукцию антиоксидантных генов (например, индукция гемоксигеназы 1 (ГО1, кодируемая геном HMOX1) через активирующий белок 1 (AP-1) [94] и рецептор γ, активируемый пероксисомным пролифератором (PPARγ) [95], или индукция глутамат-цистеинлигазы (ГЦЛ) [96] и СОД1 (источник 97) через ядерный фактор κB (NF-κB)), но оказалось, что наиболее обширную группу антиоксидантных генов (GCLC, GCLM, HMOX1, NQO1, GSTM1, GPX4, TXN и PRDX1) запускает NRF2 (источники 98, 99) (Вставка 3).
Непосредственную угрозу функционированию и структурной целостности клеток представляют •OH, ONOO— и HOX. Они слишком быстро реагируют с липидами мембран, нуклеиновыми кислотами и белками, чтобы можно было успеть эффективно их нейтрализовать низкомолекулярными ловушками свободных радикалов. К сожалению, в отношении ловушек •OH было сделано много неверных заявлений. Да, окислительный стресс включает в себя продукцию •OH, но использование экзогенных молекул для нейтрализации этих радикалов в биологических системах оказалось неэффективным. Все органические соединения реагируют с •OH с приблизительно одинаковыми константами скорости реакции, приближаясь к скорости диффузии. Таким образом, ни одно соединение не обладает большей •OH-нейтрализующей активностью, чем тысячи молекул, уже существующих в любой биологической системе. Чтобы быть эффективным хотя бы на 50%, любое соединение должно присутствовать в равной или большей концентрации, чем все эндогенные молекулы. А потому единственной эффективной стратегией избежать повреждения от •OH — это предотвратить его появление. Решения, которые могут оказаться успешными в этой стратегии — это сокращение выработки O2•, нейтрализация O2•- и H2O2. Устранение O2•- также предотвратит образование ONOO—, а устранение H2O2 предотвратит образование •OH и HOX.
Первую линию защиты против окислительного стресса формируют СОД и ферменты, обезвреживающие H2O2 и гидропероксиды липидов. Однако существуют терапевтически значимые различия между внутриклеточным пространством и внеклеточными жидкостями. Внеклеточные СОД (ВК-СОД, СОД3) обычно ассоциированы с наружной мембраной клеток и не представлены во всех внеклеточных жидкостях. Миметики СОД эффективны во внеклеточных жидкостях, где снижение продукции потенциально вредоносного ONOO— имеет дополнительное преимущество в виде сохранения ресурсов •NO, необходимого для регуляции тонуса сосудов и других важных физиологических процессов [100]. Хотя ВК-СОД связана с внешней стороной мембраны некоторых клеток, дополнительная каталазная активность большинства миметиков СОД также способствует расщеплению H2O2, чего не умеет ВК-СОД. Внутриклеточная защита включает цитозольную СОД1 и СОД2 матрикса митохондрий, которые обезвреживают O2•-, в то время как каталаза пероксисом (и митохондрий кардиомиоцитов), ГП и пероксиредоксины (PRDX) обезвреживают H2O2.
Некоторые ГП и PRDX также обезвреживают липидные пероксиды, двое из них (PRDX6 и GPX4) способны инактивировать гидропероксиды фосфолипидов. Нейтрализация O2•- малыми молекулами внутри клеток ничтожно мала по сравнению со скоростью обезвреживания эндогенными СОД, которые имеют константы скорости реакции в миллионы раз выше (~2 × 109 M-1 s-1) по сравнению с другими реакциями с O2•-.
ВК-СОД связана с наружной стороной мембраны многих клеток, и она также эффективнее большинства потенциальных “мусорщиков” O2•-. Тем не менее, миметики СОД полезны во внеклеточных средах, в которых ЕС-СОД представлена незначительно. Результатом работы СОД становится образование H2O2, что, казалось бы, не представляет большой выгоды с точки зрения антиоксидантной защиты; однако удаление O2•- предотвращает образование более опасного ONOO—, одновременно сохраняя физиологически важный •NO. Соединения с комбинированной активностью СОД и каталазы имеют преимущество перед СОД.
Вторая линия антиоксидантной защиты представляет собой синтез тиоредоксина (ТР), GCL и глутатионсинтетазу, глутатионредуктазу, и тиоредоксинредуктазу, которые используют НАДФН для восстановления GSSG и TrxS2. Следует отметить, что ферменты первой и второй линии защиты играют физиологическую роль в редокс-сигналинге и поддержании окислительно-восстановительного гомеостаза, и полное устранение H2O2 из внутриклеточной среды будет негативно влиять на клеточные функции [101].
Инактивация H2O2 и других гидропероксидов малыми молекулами очень несущественна по сравнению с работой 15 ферментов, устраняющих H2O2 и гидроперекиси липидов и 2 ферментов, обезвреживающих гидропероксиды фосфолипидов. Тем не менее, несколько миметиков ГП, включая эбселен (см. ниже), имеют константы скорости реакций, близкие к таковым у вышеупомянутых ферментов. Следовательно, соединения, пополняющие ресурсы глутатиона через цистеин (критически важной аминокислоты для синтеза глутатиона) или же просто предшественники глутатиона будут повышать эффективность эндогенной ГП или ее миметиков. Также терапевтическое преимущество будет давать увеличение синтеза глутатиона путем индукции глутаматцистеинлигазы (ГЦЛ) — лимитирующего фермента в синтезе глутатиона. Поиск соединений, которые индуцируют ГЦЛ через активацию фактора транскрипции NRF2, является одной из важных задач на протяжении уже более двух десятилетий.
Третья линия антиоксидантной защиты — восстановление поврежденных оксидантами макромолекул. Эта широкая область исследований не основная в данном обзоре; однако оксиданты индуцируют ферментные системы, которые участвуют в устранении окисленных белков [102], окисленных жирных кислот [102], ДНК, а также в репарацией ДНК [104].Вставка 3 | Сигнальный путь NRF2–EpREЯдерный фактор, родственный эритроидному фактору 2 (NRF2) относится к семейству “воротничковых” (cap‘n’collar) транскрипционных факторов, содержащих лейциновую застежку в основной области bZIP (CNC-bZIP). Впервые NRF2 был открыт Moi et al. в 1994 (источник 283) как фактор, регулирующий экспрессию β-глобина, и вскоре после этого стало ясно, что он также является активатором транскрипции NQO1, который связывается в области промотора с элементом антиоксидантного ответа (ARE) [284]. Множество исследований подробно показали, что сигнальный путь NRF2–ARE играет центральную роль в регуляции экспрессии генов антиоксидантной защиты [285]. ARE, цис-элемент связывания NRF2, более правильно бы было называть элементом электрофильного ответа (EpRE), так как “индукторы” антиоксидантов представляют из себя электрофилы: перекись водорода (H2O2), некоторые компоненты промежуточного метаболизма и соединения, образующиеся из полифенолов в пище [6].
Сигнальный путь NRF2–EpRE управляет базальной и индуцированной экспрессией более 200 генов, кодирующих белки, вовлеченные в антиоксидантную защиту, детоксикацию, апоптоз, репарацию ДНК, устранение других окисленных белков протеасомами, воспаление и другие процессы [102, 286, 287]. Роль NRF2 в индукции антиоксидантных ферментов и защиты от окислительного стресса была подтверждена на клеточных культурах и у животных с нокаутом и/или индукцией NRF2. Все больше доказательств свидетельствуют о том, что дефицит активности NRF2 снижает индукцию целевых антиоксидантных ферментов в ответ на окислительный стресс, что повышает уязвимость к окислительным повреждениям [288] и усиливает воспалительный ответ [289], в то время как повышение активности NRF2 повышает экспрессию антиоксидантных ферментов и защиту от окислительного стресса.
Молекулярный механизм и регуляция активации NRF2 в ответ на окислительный стресс обсуждались во многих недавних публикациях. Из них наиболее актуальным для терапии является недавний обзор Cuadrado и соавт. [99]. Мы же только коротко опишем регуляцию NRF2 (Рис. 3). В незадействованном состоянии большинство молекул NRF2 связаны с Kelch-подобным ECH-ассоциированным белком 1 (KEAP1) и/или с белком, содержащим β-трансдуциновые повторы (βTrCP) и быстро разрушаются 26S протеасомой после убиквитинилирования. KEAP1 является адаптором для Cullin 3-содержащего комплекса Е3 убиквитинлигазы [290], а βTrCP — субстратным рецептором для Cul1-связанной убиквитинлигазы [291]. В ответ на окислительные стимулы происходит окислительная модификация KEAP1 и он теряет способность отправлять NRF2 на деградацию. Одновременно с этим окислительное ингибирование фосфорилирования NRF2 в домене Neh6 киназой гликогенсинтазы 3β (GSK3β) останавливает взаимодействие NRF2 с βTrCP. Кроме того, NRF2 может быть активирован через аутофагическую деградацию KEAP1, опосредованную p62 (источник 292). При активации этих путей как синтезированный, так и диссоциированный (от KEAP1) NRF2 избегает деградации, после чего транслоцируется в ядро, где формирует гетеродимеры с малыми белками семейства Maf или Jun, связывается с промотором EpRE и повышает экспрессию генов-мишеней. В ядре NRF2 может быть конкурентно заингибирован BACH1 (источник 293). ChIP-seq анализ показал существенное перекрытие генов-мишеней у BACH1 (в клетках HEK293) и NRF2 (у мышиных клеток MEF) [294]. Есть доказательства, что супрессивное действие BACH1 на NRF2 может быть геноселективным. Так, инактивация BACH1 необходима для индукции HO1, но при этом не происходит с тиоредоксинредуктазой 1, хотя оба гена регулируются NRF2 (источник 295). У нокаутных по Bach1 мышей апрегулированы менее 10% генов-мишеней NRF2 [244]. Стоит сказать, что регуляция NRF2 гораздо сложнее, поскольку более простые пути, такие как NF-κB, PKC, p21, BRCA1, HRD1, CRIF1 и микроРНК вовлечены в регуляцию сигнального пути NRF2 через влияние на экспрессию NRF2, стабильность белка, активацию и транслокацию [99].
По мере выявления новых регуляторов и путей взаимодействия, становится ясно, что активность NRF2 регулируется сетью сигнальных путей, что обеспечивает ему важную роль в различных биологических процессах и ответах на различные ситуации. Пока сохраняются некоторые загадки относительно регуляции NRF2, например, непонятно, как NRF2 транспортируется в ядро и обратно, или дисрегуляция NRF2 и возникновение “потолка” его индукции при некоторых патофизиологических состояниях.
Стратегии антиоксидантной защиты
На данный момент прорабатывается несколько стратегий антиоксидантной защиты, а некоторые из них уже проходят клинические испытания. К таким стратегиям относятся инактивация O2•- до того, как он успеет прореагировать с •NO и привести к образованию ONOO— (реакция 11), и устранение H2O2 до того, как она преобразуется в •OH (реакция 7) или HOX (реакция 13); увеличение запасов восстановленного глутатиона; стимуляция синтеза антиоксидантных ферментов, особенно через активацию NRF2 (Вставка 3); ингибирование NOX (реакция 2); система антиоксидантной защиты митохондрий; пищевые добавки с антиоксидантами; и, наконец, подавление аберрантного редокс-сигналинга (вставка 2). Реакции смотрите во Вставке 1.
Миметики СОД и СОД-каталазы
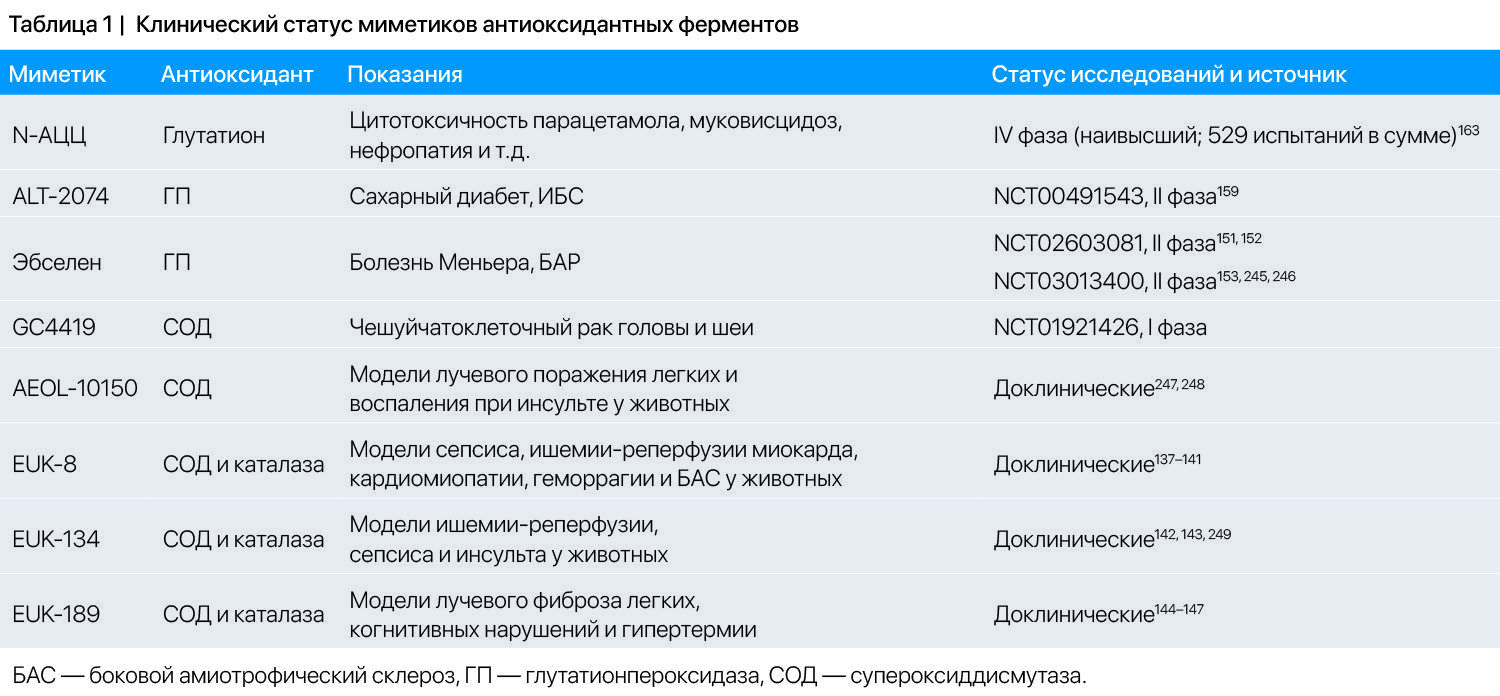
Несколько аналогов антиоксидантных ферментов уже находятся на стадии клинических исследований (Таблица 1). СОД — это единственный фермент клеток млекопитающих, который может инактивировать O2•-, и потому является ключевым компонентом антиоксидантной защиты и сохранения ресурсов •NO. Именно поэтому терапевтический потенциал СОД вызывает повышенный интерес с момента её открытия в 1969-м [105]; с того времени было разработано множество аналогов СОД. В их число входят металлопорфирины, циклические полиамины с Mn, нитроксиды, Mn-саленовые комплексы и фуллерены, химические свойства которых были описаны ранее [106,107]. Первые исследования миметиков СОД в первую очередь фокусировались на металлопорфиринах (таких как MnTM-4-PyP5+ и FeTM-4-PyP5+) [108-110]. В поздних 1990х, после выяснения зависимости между редокс свойствами металл-ассоциированного сайта и активностью СОД [111] было разработано больше порфиринов или порфириноподобных миметиков с более высокой СОД-активностью. Защитный эффект многих из этих соединений был продемонстрирован в экспериментах на животных и даже в клинических испытаниях. Но все же миметики СОД и каталазы имеют константы скорости реакции на несколько порядков ниже, чем оригинальные ферменты. Таким образом, когда они попадают в клетки, их вклад в антиоксидантную защиту оказывается незначительным. Однако, они, кажется, эффективны во внеклеточном пространстве, где концентрации антиоксидантных ферментов и субстратов очень низки или вовсе отсутствуют (Рисунок 1). Некоторые из миметиков также могут быть активны в матриксе митохондрий, но в них они одновременно могут выступать в качестве прооксидантов [112].
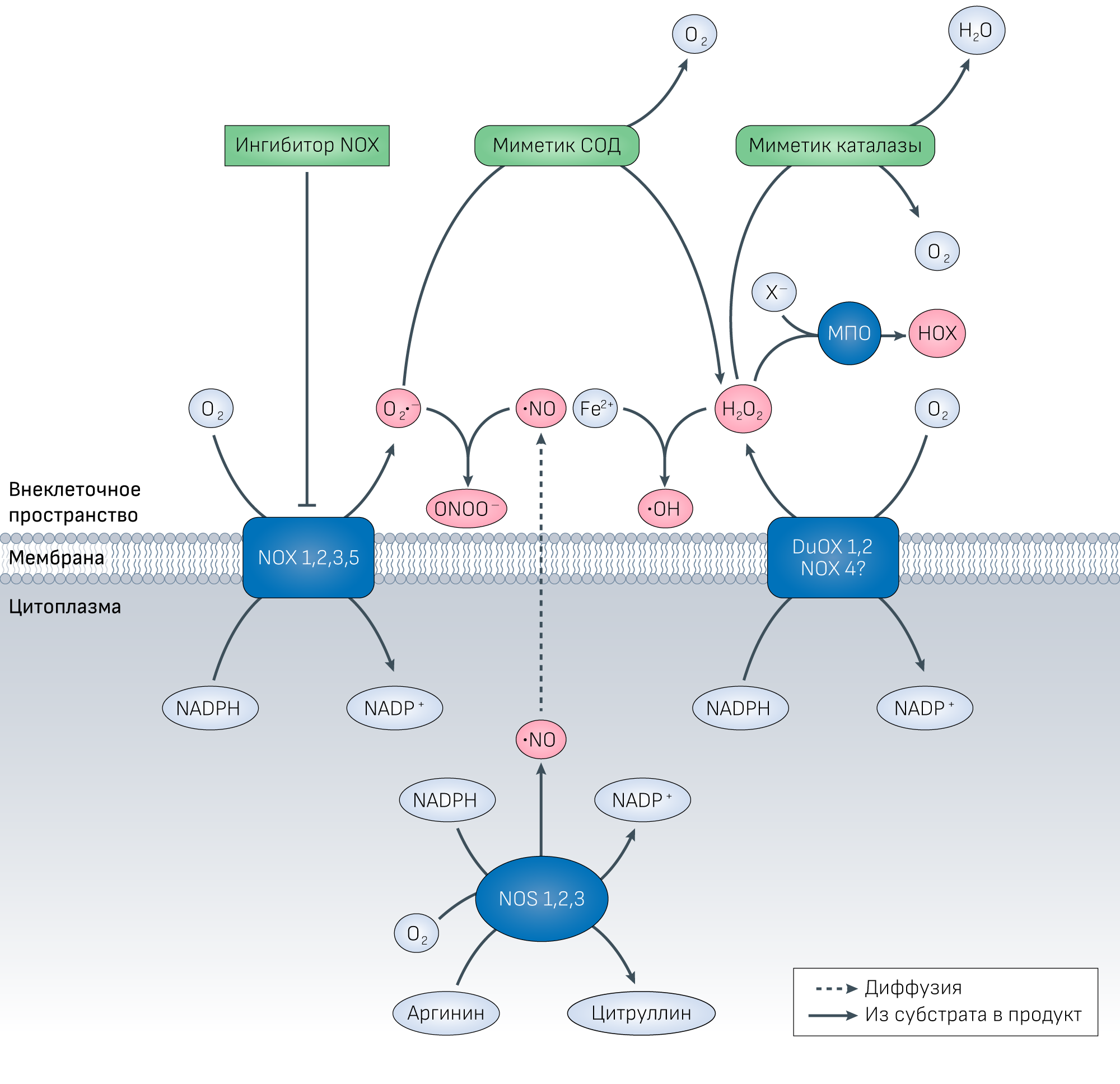
Рисунок 1 | Активные формы кислорода в межклеточном пространстве и системы защиты, обеспечиваемые миметиками СОД или каталазой, а также ингибиторами NOX.Синтез O2•− НАДН-оксидазой цитоплазматической мембраны (NOX) может быть приостановлен с помощью ее ингибиторов.
Миметики СОД катализируют реакцию дисмутации O2•− до появления из него перекиси водорода, предотвращая тем самым формирование пероксинитрита (ONOO−), что, кроме прочего, позволяет экономить оксид азота (•NO). Миметики каталазы ускоряют восстановление перекиси водорода и предотвращают синтез гипогалогеновых кислот (HOX) при участии миелопероксидазы (MPO), а также синтез гидроксил радикала (•OH) в реакции Фентона. Кажется, что каталазной активностью обладают большинство миметиков СОД. Существует еще NOX4, которая располагается внутриклеточно в мембранах органелл, но она также была обнаружена и в цитоплазматической мембране только в одном типе клеток [275], а потому ее внеклеточная функция остается под вопросом (обозначено на рисунке знаком вопроса).
NOS — NO-синтаза
Несмотря на то, что миметики СОД были разработаны для того, чтобы целенаправленно инактивировать O2•-, они неспецифичны и могут также нейтрализовать другие активные формы кислорода и азота, такие как ONOO—, гидропероксильный радикал, H2O2 и CO3•- [113,114]. Кроме того, некоторые миметики СОД, такие как Mn-порфирины, Mn(2)-циклические полиамины и M40403, могут выступать в роли прооксидантов и реагировать с тиолами [112], аскорбиновой кислотой [115] и тетрагидробиоптерином [116], таким образом оказывая влияние на редокс-чувствительные сигнальные пути и клеточную транскрипцию [117,118]. Поэтому некоторые протективные эффекты миметиков СОД могут быть связаны с их другими свойствами, кроме подражания СОД.
В поздних 1970х на основе СОД было разработано вещество орготеин, но оно не было одобрено для использования у людей [119]. Однако, проведенное двойное слепое плацебо-контролируемое исследование показало, что орготеин может быть использован для безопасной и эффективной борьбы с нежелательными эффектами лучевой терапии у пациентов с раком мочевого пузыря, например, с возникновением радиоиндуцированного острого цистита и ректита [120, 121]. Однако в другом клиническом испытании орготеин не продемонстрировал положительного влияния на лучевую терапию или острые лучевые реакции и вызвал у некоторых пациентов побочные эффекты в виде выраженной подкожной инфильтрации и покраснения в месте инъекции [122]. Сейчас орготеин используется как противовоспалительное средство у животных.
Наиболее хорошо изучен класс миметиков СОД — металлопорфирины с Mn. Уже есть данные об активности в реакции дисмутации O2•- у нескольких синтезированных вариантов порфирина с Mn [114]. Некоторые из них, такие как MnTM-2-pYp5+ и MnTE-2-pYp5+, имеют очень высокую активность. Хотя в некоторых случаях остается непонятным, ключевым механизмом является СОД-активность или какое-либо другое свойство (например, прооксидантная активность). Защитный и терапевтический эффект MnTE-2-pYp5+ и MnTDE-2-ImP5+ был показан на моделях заболеваний у животных, включая ишемический инсульт [123], лучевые поражения [124], различные виды рака [125,126], диабет [127] и повреждение сердечно-сосудистой системы [128]. Эти доклинические результаты подтверждают потенциал использования порфиринов Mn в терапии заболеваний с существенным вкладом окислительного стресса в патогенез. Соединение MnTDE-2-ImP5+ уже было безопасным в диапазоне терапевтических доз во время проведения первой фазы клинических испытаний у пациентов с БАС [129].
GC4419 — еще один многообещающий миметик СОД, представляет собой новый высокостабильный Mn(2)-содержащий пента-азамакроцикл. GC4419 избирательно реагирует с супероксид-анионом, не вступая в реакцию с другими окислителями [130]. In vitro GC4419 значительно повышал противоопухолевую токсичность AscH— [131]. Кроме того, у GC4419 были обнаружены терапевтические эффекты на моделях воспаления [132], артрита [133] и ишемии-реперфузии миокарда [134] у животных. В недавно прошедшей фазе 1 клинических испытаний GC4419 имел приемлемую безопасность у пациентов на лучевой и химиотерапии с тяжелым воспалением слизистой полости рта на фоне рака ротоглотки [135].
К другому классу СОД миметиков относятся салены — замещенные этилендиамидные комплексы металлов. Mn(3)-содержащие саленовые комплексы активны против O2•- и H2O2 [136]. Саленовые комплексы неселективны и могут реагировать с другими пероксидами и ONOO—. Типичными представителями саленов являются EUK-8, EUK-134 и EUK-189, показавшие эффективность на многих моделях заболеваний человека у животных, включая сепсис [137], ишемию-реперфузию миокарда [138], кардиомиопатию [139], геморрагический и амиотрофический боковой склероз (EUK-8) [141], ишемия-реперфузия почки [142] и инсульт (EUK-134) [143]; радиационный фиброз легких [144], когнитивные нарушения [145], дисфункцию диафрагмы при монокроталин-индуцированной легочной гипертензии [146] и гипертермии (EUK-189) [147]. Салены пока не проходили ни одного клинического испытания.
Миметики глутатионпероксидазы (ГП)
К текущему моменту синтезированы различные миметики ГП [148], среди которых наиболее известно селенорганическое соединение эбселен (2-фенил-1,2-бензизоселеназол-3(2H)-1) с широкой специфичностью для субстратов — от H2O2 и малых органических гидропероксидов до мембраносвязанных фосфолипидов и гидропероксидов холестерола [149]. Эбселен также способен индуцировать ферменты 2й фазы биотрансформации [150]. Исследования на животных показали, что эбселен способен снижать окислительное повреждение [150], предотвращать острую потерю наружных волосковых клеток [151] и ухудшение слуха, а также подавлять воспаление [152]. Соответственно, было проведено несколько клинических испытаний при таких заболеваниях, как болезнь Меньера (III фаза, NCT04677972), биполярное аффективное расстройство [153], полная окклюзия средней мозговой артерии [154], отсроченный неврологический дефицит после аневризматического субарахноидального кровоизлияния [155] и острого ишемического инсульта [156]. В этих исследованиях эбселен хорошо переносился при приеме внутрь, имел хорошую биодоступность и терапевтическое действие.
ALT-2074 (BXT-51072) — новый аналог эбселена с повышенной активностью и эффективностью. ALT-2074 подавлял воспалительную реакцию в эндотелиальных клетках in vitro [157], уменьшал окислительное повреждение и гибель нейронов [158], а на модели ишемии-реперфузии сердца у мышей уменьшал размер инфаркта [159]. Завершено клиническое испытание II фазы ALT-2074 (NCT00491543) при диабете и ишемической болезни сердца, но данные еще не опубликованы. Клиническое испытание с участием больных псориазом (NCT00782613), было прекращено, но причины этого остаются неизвестными.
Хелатирование железа
При высвобождении из белков ионы железа и меди могут участвовать в синтезе •OH, при этом одни хелаторы ускоряют эти реакции, а другие ингибируют. В принципе, использование ингибирующих хелаторов было бы отличной стратегией для предотвращения синтеза •OH; однако, поскольку железо необходимо для многих биологических процессов, использование хелаторов в терапии обычно ограничивается профилактикой перегрузки железом у пациентов с серповидно-клеточной анемией и талассемией, которым требуются частые переливания крови [161].
Повышение уровня глутатиона
Хотя в большинстве клеток концентрация глутатиона находится в миллимолярном диапазоне, она, как правило, значительно падает в результате окислительного стресса. Таким образом, подходы, направленные на поддержание или восполнение запасов глутатиона, реализуются с помощью его предшественника, цистеина — лимитирующей аминокислоты в синтезе глутатиона. Такие вмешательства показали эффективность при различных заболеваниях.
N-ацетилцистеин
N-ацетилцистеин (АЦЦ) является одним из наиболее изученных терапевтических антиоксидантов (Таблица 1). Он растворим в воде и быстро всасывается в основном через мембранный белок анионного обмена [162]. В клетках АЦЦ деацетилируется с образованием цистеина, а значит антиоксидантный эффект АЦЦ в основном опосредован через пополнение ресурсов глутатиона [163]. NAC также может уменьшать количество цистеиновых конъюгатов в плазме крови [162]. АЦЦ используется для лечения многих патологий, включая повреждение печени при отравлении парацетамолом (ацетаминофен по номенклатуре USAN) [164], муковисцидоз, при котором он доставляется ингаляционно [165] и нефропатию [166]. В исследованиях на животных и в клинических испытаниях АЦЦ изучается для профилактики или лечения многих других заболеваний и состояний. Результаты этих исследований противоречивы и пока нет консенсуса. Неэффективность АЦЦ в ряде случаев может быть обусловлена тем, что окислительный стресс выступает лишь вторичным фактором развития заболевания.
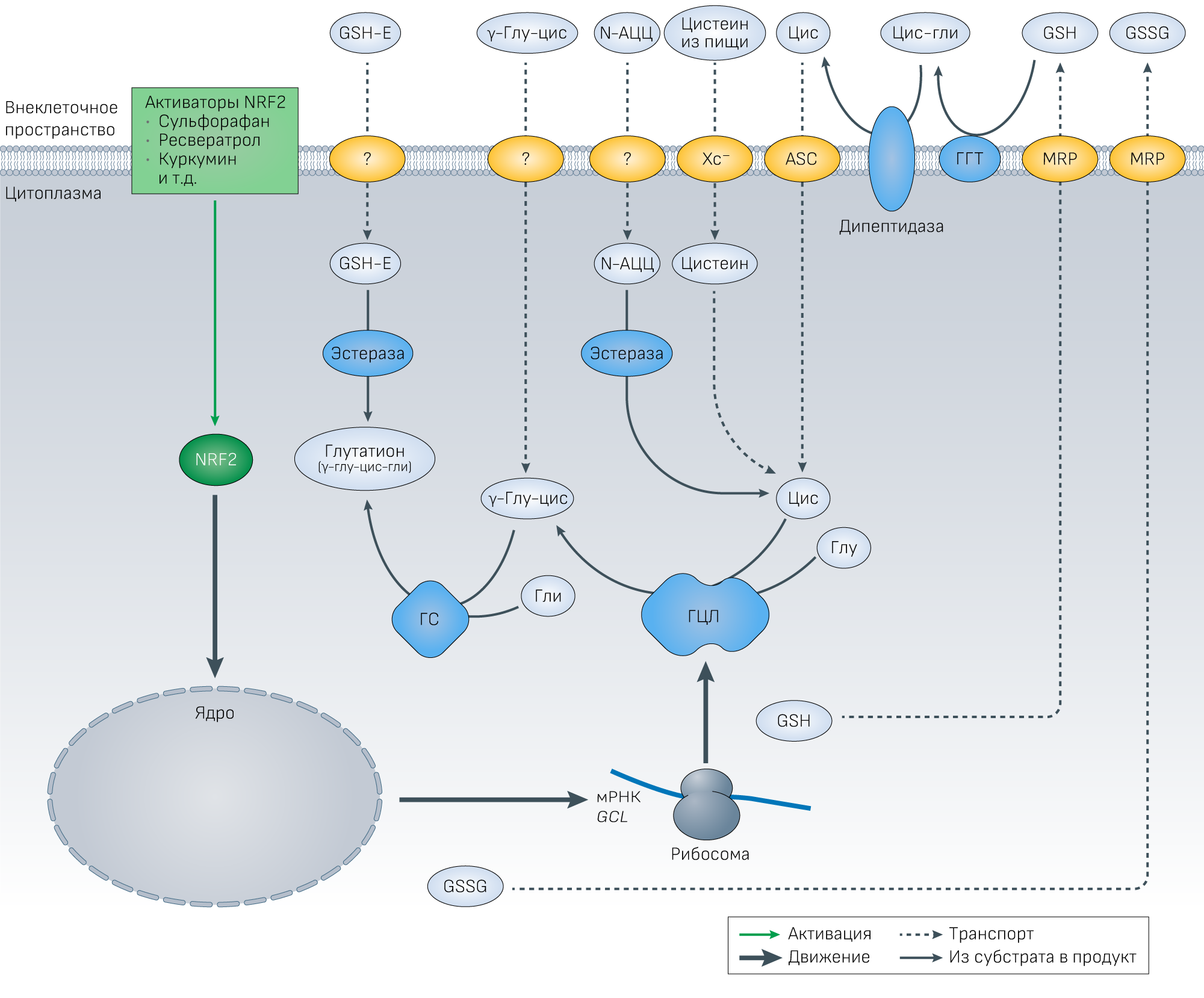
Рисунок 2 | Метаболизм глутатиона и стратегии по увеличению уровня глутатиона.Глутатион синтезируется при участии глутамат-цистеинлигазы (ГЦЛ) и глутатионсинтазы, где ГЦЛ является лимитирующим ферментом, а скорость данной реакции определяет доступность цистеина. Восстановленный глутатион и дисульфид глутатиона (GSSG) экспортируются за пределы клетки при участии белка множественной лекарственной устойчивости (MRP), где внеклеточный глутатион последовательно метаболизируется мембраносвязанной гамма-глутамил транспептидазой (ГГТ) до цистеинилглицина и гамма-глутамила, и дипептидазой до цистеина и глицина. Эти аминокислоты транспортируются обратно в клетки и снова участвуют в синтезе глутатиона. N-ацетилцистеин (N-АЦЦ) деацитилируется эстеразой до цистеина, а эфиры глутатиона прямо превращаются эстеразой в глутатион. Гамма-глутамилцистеин (γ-глу-цис) позволяет обойти скорость-лимитирующую реакцию синтеза глутатиона. Электрофилы вызывают активацию NRF2, которая управляет транскрипцией двух субъединиц ГЦЛ и глутатион-синтазы.
Были определены следующие транспортеры:
ASC — Na-зависимый аланин-серин-цистеиновый транспортер; Xc− — система антипорта цистеин/глутамат.
Знак вопроса обозначает неустановленные транспортеры/каналы для эфиров глутатиона, γ-глу-цис и АЦЦ.
Эфиры глутатиона
Сам глутатион плохо проникает в большинство клеток, а экзогенно введенный глутатион быстро разрушается в плазме [167]. Таким образом, решение представляется в виде доставки пролекарства в виде модифицированного глутатиона. На предмет эффективности доставки глутатиона были изучены его эфирные производные, включая монометиловый (GSH-OMe), моноэтиловый (GSH-MEE), диэтиловый (GSH-DEE) и изопропиловый эфиры. В GSH-MEE этерифицирована карбоксильная группа остатка глицина (Glu-Cys-Gly-OEt), тогда как в GSH-DEE этерифицированы остатки глутамата и глицина (tEO-Glu-CysGly-OEt). Эфиры глутатиона липофильны, более эффективно транспортируются через клеточную мембрану и устойчивы к разрушению γ-глутамилтранспептидазой в плазме [168]. Попав внутрь клетки, эфиры глутатиона быстро гидролизуются неспецифическими эстеразами и образуют глутатион. Транспорт GSH-DEE в клетки более эффективен по сравнению с моноэфиром [169], а клетки человека могут быстро превращать диэтиловый эфир в моноэфир, который затем гидролизуется в глутатион.
Высокая эффективность эфиров глутатиона в повышении уровня тканевого и/или внутриклеточного глутатиона была доказана во многих исследованиях на клеточных культурах и животных [170-175]. Подкожное или внутрибрюшинное введение эфиров глутатиона животным повышало уровень глутатиона в различных тканях, включая печень [170], почки [170], селезенку, поджелудочную железу и сердце [176], но не мозг [177]. Уровень глутатиона в мозге может быть увеличен путем введения GSH-MEE [177] в желудочки мозга [174]. Пероральный прием также может повысить уровень глутатиона в тканях, но менее эффективно [176].
Относительная эффективность различных эфиров глутатиона по его доставке в ткани остается неясной из-за ограниченного количества данных. Некоторые исследования на клеточных культурах показывают, что GSH-DEE более эффективен в доставке глутатиона, чем GSH-MEE [169]. GSH-DEE по-разному метаболизируется в плазме крови животных и людей. У мышей и крыс GSH-DEE в плазме быстро превращается в GSH-MEE под действием α-эстеразы, тогда как в плазме человека (и многих других видов, включая хомяка, морскую свинку, кролика и овцу) активность α-эстеразы плазмы отсутствует — это означает, что GSH-DEE может проникать в ткани более эффективно, чем GSH-MEE [169].
Однако пока не проведено ни одного прямого сравнения относительной эффективности различных эфиров глутатиона в клинических исследованиях. Приведенные выше данные свидетельствуют о том, что люди, по-видимому, переносят препараты глутатиона без побочных эффектов. Также эфиры глутатиона как пролекарства эффективно доставляли его в ткани и уменьшали окислительное повреждение в клетках у животных, но клинические испытания эфиров глутатиона не проводились. На рисунке 2 обобщены стратегии поддержания уровня глутатиона в клетках.
Активаторы NRF2
При многих заболеваниях, связанных с развитием окислительного стресса, в том числе сердечно-сосудистых [178], нейродегенеративных [179] и заболеваниях легких [180], происходит нарушение сигналинга NRF2 (Вставка 3, Рисунок 3). Более того, известно, что активаторы NRF2 — потенциальные соединения для стимуляции антиоксидантной защиты и лечения патологий. Индуцирование антиоксидантных ферментов, особенно через NRF2 — одно из главных направлений развития антиоксидантной терапии. Действительно, в тех случаях, когда малые молекулы типа полифенолов оказываются эффективными, они действуют преимущественно путем индуцирования ферментов антиоксидантной защиты через сигнальный путь NRF2 [6]. В зависимости от механизма действия активаторы NRF2 разделяются на 5 групп (Рисунок 3):
- модификация Kelch-подобного ECH-связанного белка 1 (KEAP1; регулирует протеосомную деградацию NRF2), который инактивируется, когда его цистеины образуют аддукты с электрофилами, или когда они окисляются с образованием дисульфидных мостиков;
- нарушение взаимодействия между белком, содержащим β-трансдуциновый повтор (βTrCP; убикситинилирует NRF2, после чего тот отправляется на деградацию) и NRF2. Осуществляется через окислительное ингибирование киназы гликогенсинтазы 3β (GSK3β), после чего не происходит фосфорилирование NRF2 в домене Neh6 не происходит и взаимодействие с βTrCP нарушается;
- секвестрация KEAP1 при помощи p62;
- de novo синтез NRF2, который избегает деградации инактивированным KEAP1 (источник 181);
- ингибиторы BACH1, в результате их работы уменьшается супрессивное влияние BACH1 на эффекты NRF2. Подход включает в себя ингибиторы трансляции BACH1 [182] и стимуляцию разрушения BACH1 [183].
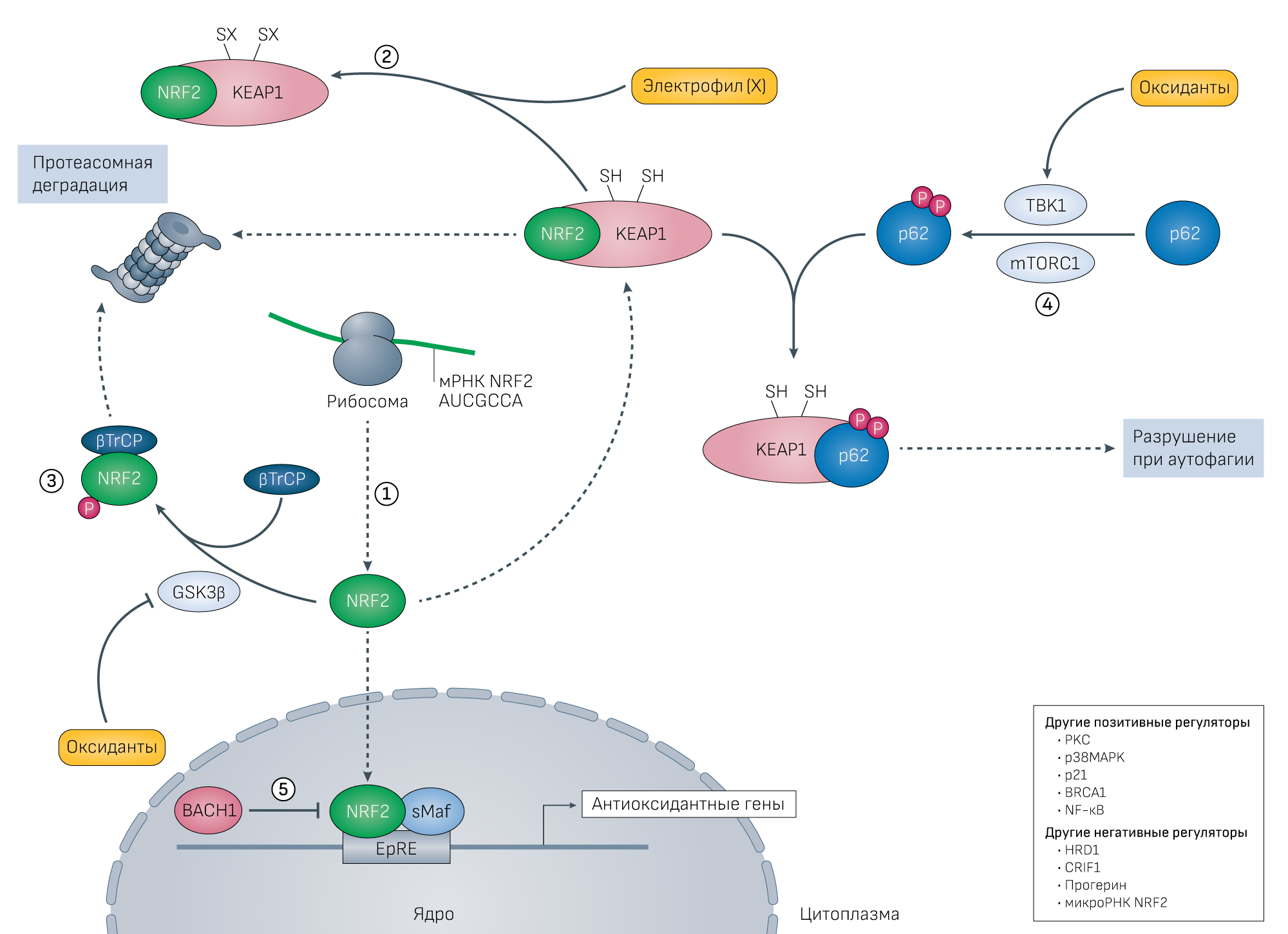
Рисунок 3 | Сигнальный путь NRF2 и терапевтические подходы с использованием антиоксидантов.1. Транскрипционный фактор NRF2 постоянно синтезируется в клетках, но в покое его транспорт в ядро ограничен. Это связано с его деградацией при связывании с Kelch-подобным ECH-связанным белком 1 (KEAP1), который ускоряет деградацию NRF2 при участии 26S-протеасомы. Антиоксидантный подход здесь заключается в усилении синтеза NRF2.
2. Под воздействием электрофилов происходит алкилирование KEAP1 и он теряет способность ускорять разрушение NRF2. Следовательно, другой подход состоит в использовании нетоксичных электрофилов, способных вызывать алкилирование KEAP1. На рисунке у KEAP1 SH обозначает тиол, а SX — аддукт с электрофилом (X).
3. Параллельно киназа гликогенсинтазы 3β (GSK3β) может фосфорилировать NRF2, после чего он вместе с белком, содержащим β-трансдуциновый повтор (βTrCP), аналогично разрушается в протеасомах. Этот процесс ингибируется путем окислительной инактивации GSK3β. В результате окислительного ингибирования GSK3β не происходит фосфорилирования NRF2 в домене Deh6 и взаимодействие NRF2 и βTrCP нарушается. Таким образом, еще один подход заключается в ингибировании GSK3β.
4. Окислительная деградация KEAP1 происходит через p62-опосредованную секвестрацию и аутофагию, этот процесс запускается фосфорилированием p62 при участии TANK-связывающей киназы 1 (TBK1) и механистической мишени комплекса рапамицина 1 (mTORC1). Поэтому p62 может служить потенциальной мишенью. Синтезированные молекулы NRF2, которые избежали разрушения, транслоцируются в ядро и связываются с последовательностями EpRE в области промоторов антиоксидантных генов и усиливают их экспрессию. Активность NRF2 также может быть повышена через фосфорилирование протеинкиназой С (PKC) [269] и его взаимодействие с другими белками, такими как p21 ( источник 270) и BRCA1 (источник 271).
5. В ядре активность NRF2 подавляет BACH1, конкурируя за образование гетеродимеров с малыми белками Maf (sMaf) или Jun и связываясь с элементом электрофильного ответа (EpRE) [272, 273, 274]. Таким образом, соединения, ингибирующие BACH1, предлагают альтернативный терапевтический подход для повышения экспрессии некоторых NRF2-регулируемых генов. К другим негативным регуляторам NRF2, представляющим собой потенциальные терапевтические мишени, относятся HRD1, CRIF1, прогерин и микроРНК для NRF2 (источник 99).
Экстракты чая, кокоса и многих овощей и фруктов, включая брокколи, ростки брокколи, виноградные косточки и куркуму, могут активировать сигнальный путь NRF2 и индуцировать антиоксидантные ферменты [184, 185]. Некоторые из них сейчас проходят клинические испытания в качестве средств лечения и профилактики, к примеру, ХОБЛ, остеоартрита, тугоподвижности суставов и диабетической нефропатии (www.clinicaltrials.gov). Yagishita и соавт. [186] суммировали текущий прогресс касаемо брокколи/ростков брокколи, в том числе лекарственные формы, их биодоступности, эффективности и дозы в клинических испытаниях. Они пронаблюдали некоторые благоприятные эффекты, включая усиление антиоксидантной способности, однако для разработки и валидирования биомаркеров фармакодинамики в организме человека требуется больше усилий. Как было отмечено выше, усиление антиоксидантной защиты может быть ограничено при лечении и профилактике заболеваний, если окислительный стресс вторичен по отношению к основной патологии. Основной механизм антиоксидантного эффекта пищевых добавок, в частности, кумаринов и полифенолов из фруктов и овощей, заключается в их окислении в электрофильные хиноны, которые затем формируют аддукты с цистеинами KEAP1 [6].
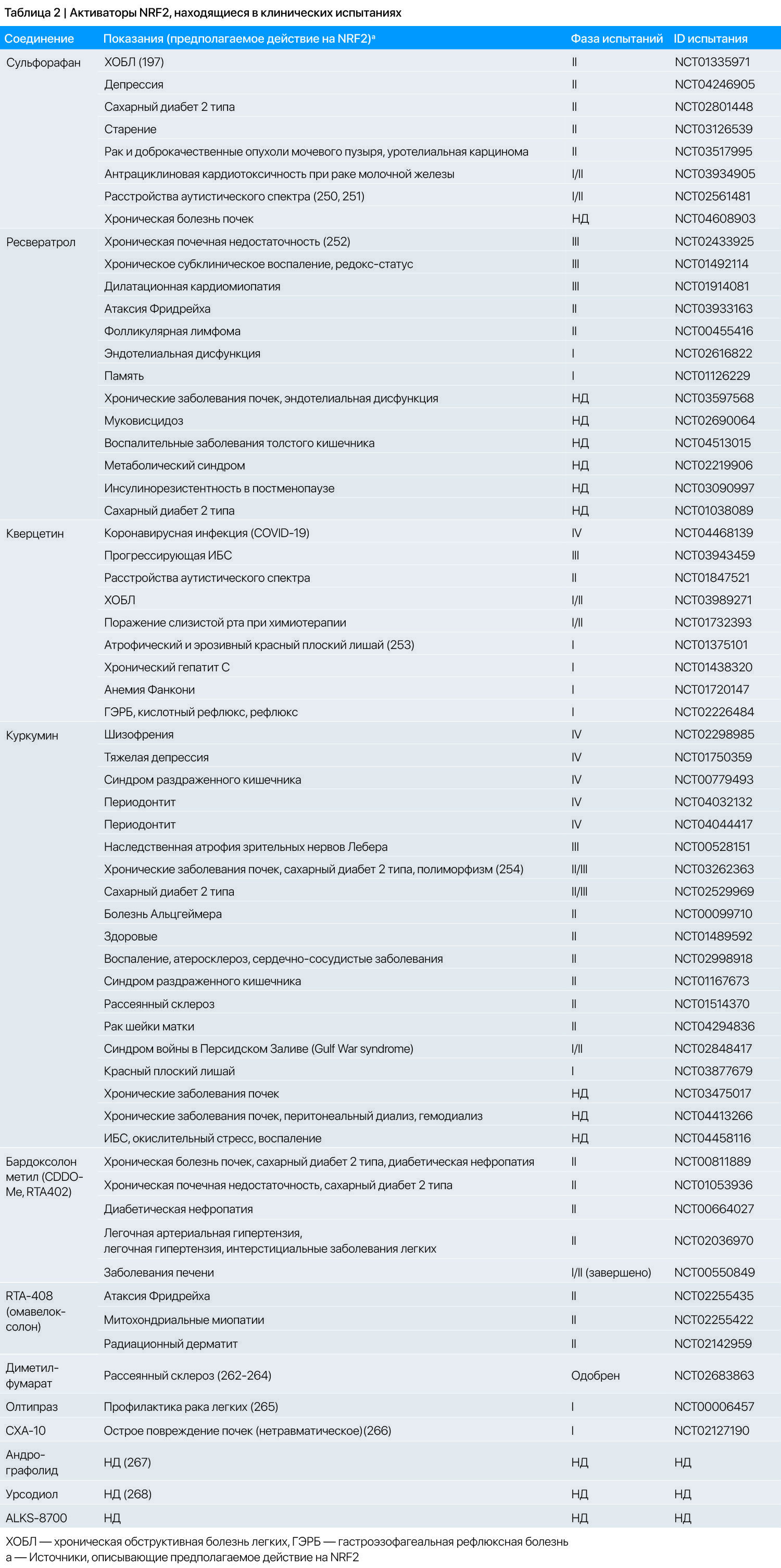
Эффективность многих из этих активаторов NRF2 в качестве индукторов антиоксидантных ферментов подтверждена во многих исследованиях на животных. Несколько активаторов NRF2 из пищевых источников, включая куркумин, сульфорафан и ресвератрол, производятся в качестве пищевых добавок, в то время как другие активаторы NRF2 исследуются как лекарства в клинических испытаниях [187]. В источнике 187 также описаны отдельные электрофильные активаторы NRF2 и соответствующие клинические испытания. Отмечается, что эти активаторы NRF2 имеют противовоспалительные свойства [188-190], часть из которых реализуются независимо от NRF2. В таблице 2 перечислено суммарное число клинических испытаний отдельных активаторов NRF2 пищевого происхождения и отмечены те, чьи эффекты основаны на активации NRF2 и/или антиоксидантном потенциале. Следует отметить, что возможна ситуация, когда некоторые соединения, у которых не изучалась активация NRF2, не указаны, хотя они на самом деле активируют NRF2.
Трудности на пути фармакологической активации NRF2
Существует ряд проблем и трудностей, связанных с использованием активаторов NRF2 в терапии [191, 192]. Первая касается низкой эффективной биологической концентрации, так как большинство активаторов NRF2 являются электрофилами и быстро метаболизируются, поэтому их биодоступность для определенных органов и тканей может оказаться слишком низкой. Однако, есть некоторые доказательства обратимости аддуктов Михаэлиса нуклеофилов (включая цистеины KEAP1) с такими электрофилами как цианоеноны [193], и это может существенно увеличить их биодоступность и концентрацию in vivo. Это предположение было проверено при помощи синтетического цианоенона TBE31, который имел период полувыведения из крови 10 часов [194] и значительно повышал активность NRF2 in vivo в наномолярных концентрациях [195]. Остается неясным, имеется ли подобная обратимость ковалентных аддуктов у других электрофилов, особенно природных соединений типа сульфорафана и куркумина. Стоит сказать, что в клинических испытаниях существует противоречие относительно эффективности перорального сульфорафана. Сообщалось как о повышении экспрессии антиоксидантов [196], так и об отсутствии эффекта [197].
Другой ключевой проблемой является риск неспецифичности действия. Кроме активации NRF2 и индукции антиоксидантных ферментов, некоторые активаторы NRF2 могут затрагивать другие сигнальные пути и биологические процессы. Например, сульфорафан может подавлять воспалительные реакции через ингибирование NF-κB [188] и активацию инфламмасом [198], а также вызывать остановку клеточного цикла, ингибируя сигнальные пути PI3K–AKT и MAPK–ERK [199]. Большинство этих эффектов были обнаружены in vitro на клеточных культурах с концентрациями сульфорафана >10 μM, которых очень сложно достичь in vivo. Понимание NRF2-независимых эффектов важно для выяснения механизмов благоприятных и терапевтических эффектов, хотя для большинства активаторов NRF2 это пока не было тщательно изучено, особенно если дело касается зависимости доза-эффект in vivo.
Активация NRF2 и индукция антиоксидантов неспецифичны и не ограничиваются одним типом клеток или органом, что может приводить к побочным эффектам. Например, есть данные, что активация NRF2 может как предотвращать, так и провоцировать развитие рака [200-202]. В литературе обсуждается [207-209], что в исследованиях на клеточных культурах повышенная активность NRF2 способствовала резистентности к противоопухолевым препаратам [203-206]. Для окончательных выводов данных пока недостаточно, и для выяснения роли NRF2 в канцерогенезе и резистентности к химиотерапии необходимы более системные исследования in vivo. Если повышенная активность NRF2 действительно способствует росту опухоли и/или повышает резистентность к химиотерапии, то следует избегать системного введения активаторов NRF2 по крайней мере у групп риска, включая онкологических больных на химиотерапии. Другие побочные эффекты длительной активации NRF2 менее изучены. Было предложено несколько стратегий для предотвращения системных побочных эффектов, включая разработку неэлектрофильных препаратов и препаратов, которые работают только в местах с активным окислительным стрессом.
Ингибирование NADPH оксидазы (NOX)
Как источник O2•- и H2O2 — NOX играет важную роль в редокс-сигналинге и уничтожении микроорганизмов, но ее чрезмерная активация может привести к повреждению нормальных тканей. Известно два типа ингибиторов NOX — прямые ингибиторы ферментативной активности и ингибиторы сборки мультибелкового комплекса NOX2. Из препаратов первого типа в исследованиях широко используется хлорид дифенилениодония (DPI), который неспецифически ингибирует флавопротеины, а также блокирует транспорт йода [210]. У нескольких ингибиторов NOX, таких как эбселен, CYR5099, апоцинин и GKT137831, эффективных в доклинических и клинических испытаниях, были обнаружены свойства, не связанные с ингибированием NOX [211]. Тем не менее, потенциальная ценность ингибирования NOX1, NOX2 и NOX4 был показана в исследованиях на животных с делецией гена [212], и поиск низкомолекулярных ингибиторов NOX продолжается.
Терапевтическое потенциалом обладают нарушающие сборку NOX-комплексов короткие пептиды [213]. Хотя эти малые пептиды могут быть более специфичны в отношении NOX, чем ингибиторы активного сайта, ни один из них не продвинулся до клинических испытаний. Третий потенциальный подход — вмешательство в синтез компонентов NOX, однако и здесь все находится на стадии доклинических исследований.
Антиоксидантные защиты митохондрий
Утечки электронов из дыхательной цепи митохондрий приводят к синтезу O2•-. Хотя подавление продукции O2•- возможно либо путем повышения уровня разобщающих белков, либо торможением потока электронов, сопутствующее уменьшение синтеза АТФ делает этот подход малоперспективным. Тем не менее, эта стратегия была предложена в качестве предотвращения повреждения от гипергликемии при сахарном диабете [214]. В качестве специфического ингибитора продукции O2•- комплексом I ЦПЭ было предложено вещество OP2113, так как оно не нарушает продукции АТФ и может использоваться у людей [215]. Однако, препарат еще не изучался в клинических испытаниях.
Как обсуждалось ранее, увеличение СОД2 приводит к усилению синтеза H2O2 в митохондриях за счет реакции 1 (QH•- + O2 ↔ Q + O2•-) (Вставка 1) путем дисмутации O2•-. Таким образом, миметики СОД, проникающие в митохондрии, будут способствовать продукции H2O2. Однако, так как они обладают еще и каталазной активностью, они, вероятно, будут оказывать благотворный эффект [216], тормозя образование OONO— и защищая белки с железо-серными кластерами. Проникать в митохондрии умеет и эбселен, но он может провоцировать нежелательные токсические эффекты [217]. Стоит отметить, что большой отрицательный заряд внутренней мембраны митохондрии делает ее приятной мишенью для антиоксидантов и миметиков антиоксидантов путем присоединения к ним липофильных катионов [218]. Эта область исследований находится пока на ранних этапах развития, но использует в своей основе тех же принципы антиоксидантной защиты, которые были описаны в этом обзоре ранее.
Антиоксиданты из пищевых источников
Наиболее используемые и изученные пищевые антиоксиданты — это аскорбиновая кислота (витамин С) и альфа-токоферол (витамин Е). Другие пищевые добавки, включая селен, рибофлавин и некоторые металлы, являются незаменимыми кофакторами антиоксидантных ферментов. Их адекватное поступление необходимо для того, чтобы индукторы этих ферментов достигли своих наиболее эффективных уровней активности, но их обсуждение выходит за рамки данного обзора. Витамин С относится к водорастворимым витаминам и не может синтезироваться в организме человека, а потому должен поступать в качестве необходимой пищевой добавки. Витамин С нужен для синтеза коллагена, а также других белков и молекул [219]. Как важный антиоксидант [220] он выступает в качестве донора электронов и нейтрализует таким образом свободные радикалы. Жирорастворимый витамин Е находится в цитоплазматической мембране и участвует во многих биологических процессах. Функции и механизмы действия витамина Е представляют большой интерес даже спустя 100 лет после открытия. Важность антиоксидантной роли витамина Е была неоднократно продемонстрирована [221–223] в условиях окислительного стресса или на фоне дефицита других антиоксидантных систем [223, 224]. Витамин Е восстанавливает пероксильные радикалы с формированием токопероксил радикала, который затем восстанавливается при участии витамина С. Таким образом, витамин Е помогает поддерживать целостность длинноцепочечных полиненасыщенных жирных кислот мембраны, а также регулирует биоактивность ее компонентов и сигнальные пути, связанные с мембранными липидами.
Алиментарный дефицит витаминов Е и С редко встречается у здоровых людей, они получают их достаточное количество с пищей. При недостаточном или несбалансированном питании, а также при некоторых заболеваниях [225, 226], может потребоваться добавка витаминов Е и С в рацион. Поскольку эти витамины являются антиоксидантами, крайне интересен их терапевтический потенциал. В ходе многочисленных исследований и клинических испытаний было установлено, что витамины С и Е оказывают благотворное влияние на снижение частоты различных заболеваний, многие из которых, вероятно, связаны с окислительным стрессом, включая рак, сердечно-сосудистые заболевания и катаракту [227]. Однако эти данные противоречивы, поскольку почти такое же количество исследований не выявило значимого эффекта. Предполагалось, что витамин С и витамин Е обладают низкой токсичностью и не вызывают серьезных побочных эффектов при гораздо большем потреблении, чем это необходимо для выполнения их функции витаминов. Однако в ряде исследований на животных было показано, что антиоксидантные добавки, включая N-АЦЦ, витамин Е и растворимый аналог витамина Е Тролокс, способствуют развитию и метастазированию рака, например, опухолей легких, меланомы и кишечника на мышиных моделях [228-230]. А потому потенциальное влияние антиоксидантов на развитие рака, включая вышеупомянутые активаторы NRF2, вызывает серьезные опасения в отношении использования антиоксидантных добавок, и для решения проблемы такого двойственного действия антиоксидантов необходимы новые стратегии.
Ингибирование аберрантного редокс-сигналинга
В ранние годы изучения редокс-биологии акцент практически полностью делался на повреждения, вызванные оксидантами. Хотя исследования демонстрировали, что добавление нелетальных доз H2O2 или других оксидантов способно стимулировать сигнальные пути, только в середине 1990-х годов впервые была отмечена активация NF-κB путем эндогенного синтеза H2O2 [231]. К концу 1990-х годов Ламбет и соавторы [232] описали семейство NOX из семи изоферментов и начали изучать их связь с сигнальными путями. В настоящее время редокс-сигналинг является основным направлением этой области, однако широкое освещение этой темы выходит за рамки данного обзора. Читатель может посмотреть соответствующие обзоры по теме [4, 233]. Тем не менее, как уже говорилось ранее, H2O2 является основным вторичным мессенджером редокс-сигналинга и, как и в случае с другими вторичными мессенджерами, дисрегуляция его продукции может приводить к аберрантному сигналингу [233]. Предотвратить дисрегуляцию сложно, поскольку попытки подавить продукцию оксидантов белками NOX или митохондриями, как описано в предыдущих разделах, могут нарушить физиологически важные сигнальные пути, в том числе регуляцию синтеза лейкотриенов и простагландинов, для которых необходим низкий уровень H2O2 или гидроперекисей липидов [234].
Более успешным подходом может быть вмешательство в специфические для разных токсических воздействий пути редокс-сигналинга. Для иллюстрации этого подхода мы приводим один пример [235]. Загрязнение воздуха предполагает содержание в нем частиц различного состава, в том числе силикаты с молекулами железа на поверхности. Такие частицы могут активировать сигнальный путь NF-κB в макрофагах, который можно ингибировать миметиками СОД и/или каталазы, но также можно вмешаться в сигнальные пути, запускаемые железо-опосредованным перекисным окислением липидов, которое приводит к разрушению липидных рафтов и последующей активации фосфохолин-специфической фосфолипазы C (PC-PLC). Трициклодекан-9-ил ксантан (D609) — ингибитор этого фермента — ранее был безуспешно испробован в качестве противоопухолевого агента, однако он останавливал NF-κB-опосредованную продукцию цитокинов, вызванную частицами загрязненного воздуха. Таким образом, D609 выступает примером соединения, которое не является антиоксидантом, но подавляет аберрантный сигналинг, вызванный оксидантами. Интересно, что D609 влияет на сигнальный путь PC-PLC при инициации последнего эндотоксином [236], что не связано с редокс-сигналингом. Существует бесчисленное множество соединений с аналогичным потенциалом ингибирования аберрантного сигналинга, хотя они и не являются специфичными для редокс-сигналинга.
Вызовы и ограничения в борьбе с окислительным стрессом
Окислительный стресс — компонент патогенеза многих заболеваний и токсикоинфекций, и представленные выше антиоксидантные средства защиты открывают важные направления по предотвращению или уменьшению патологии. Тем не менее, существуют ограничения, которые ставят под сомнение наши возможности терапевтического использования антиоксидантных стратегий.
Роль окислительного стресса в патогенезе
Эффективность антиоксидантной защиты ограничена степенью участия окислительного стресса в развитии патологии. Если окислительный стресс вторичен в патогенезе заболевания, что бывает чаще, чем когда он выступает основной причиной, то предотвращение окислительного стресса может не оказать существенного влияния на прогрессирование заболевания. На самом деле, это одна из основных причин того, что антиоксиданты практически не влияют на патологию, даже если они явно повышают антиоксидантную защиту и снижают маркеры окислительного стресса. Это ограничение, возможно, является самым важным фактором, который упускают из виду при оценке эффективности антиоксидантной защиты в клинических исследованиях. Необходимо определить, если антиоксиданты не влияют на основную причину заболевания, то в какой степени они могут облегчить отдельные симптомы. Коммерциализация продуктов, содержащих малые молекулы, которые являются антиоксидантами химически, но не функционируют в качестве таковых in vivo, в конечном итоге не сможет продемонстрировать пользу, превышающую ту, которую могут принести низкомолекулярные индукторы антиоксидантных ферментов, присутствующие в адекватном рационе питания. Это разочарование усложнит проблему разработки и получения общественного признания действительно эффективных терапевтических подходов.
Фармакокинетика низкомолекулярных ловушек свободных радикалов
Ключевое ограничение антиоксидантной защиты — это пренебрежимо малый эффект нейтрализации свободных радикалов малыми молекулами. Заявления о том, что какой-либо антиоксидант является ловушкой для •OH, бессмысленны, так как почти все малые молекулы реагируют с •OH с приблизительно равной скоростью. Таким образом, единственной защитой от •OH служит предотвращение его образования, а наиболее эффективным способом является уменьшение уровня H2O2. Для нейтрализации O2•- внутри клеток придется конкурировать с уже распространенной и высокоактивной СОД, катализирующей реакцию 3 ((2O2•- + 2H+ → H2O2 + O2) (Вставка 1)) со скоростью как минимум в 105 раз выше, чем любая другая реакция с O2•-, за исключением •NO [237]. Аналогично 15 ферментов, нейтрализующих H2O2 в реакциях 4-6 (2H2O2→2H2O + O2; H2O2 + 2Trx(SH)2 → TrxS2 + 2H2O; H2O2 + 2GSH→GSSG + 2H2O) (Вставка 1), превзойдут по внутриклеточной активности большинство агентов. Таким образом, по фармакокинетическим соображениям фактически исключена возможность использования ловушек свободных радикалов для эффективной антиоксидантной защиты внутри клеток [6]. Однако внеклеточно миметики СОД и каталазы с относительно высокими константами скорости реакций (по сравнению с неферментативными реакциями O2•- и H2O2) могут оказаться эффективны. Они, конечно, не настолько эффективны, как эндогенные СОД и каталаза, но их константы скорости примерно в 105 раз выше, чем для большинства цистеинов в составе белков. Миметики СОД также могут накапливаться в высоких концентрациях в митохондриальном матриксе, связываясь липофильным катионными группировками, и могут быть эффективны в микроокружении [106], где гиперэкспрессия эндогенной СОД2 повышает продукцию H2O2 [238]. Однако долговременные последствия повышения митохондриальной активности СОД выше физиологических значений пока не изучены.
Витамин Е является исключением среди низкомолекулярных ловушек свободных радикалов, поскольку он сравнительно быстро вступает в реакцию с гидроперекисными радикалами липидов, а также концентрируется в мембранах. Тем не менее, антиоксидантные подходы, которые казались рабочими в культурах клеток и на моделях животных, часто не достигали значимых эффектов в клинических испытаниях. Основной причиной такого расхождения является огромная разница в соотношении экзогенных агентов in vitro и in vivo [6]. Объяснением может быть то, что в лабораторном корме наблюдается дефицит витамина Е и селена [239], что создает ситуацию, в которой антиоксиданты выполняют работу по восстановлению редокс-гомеостаза, и таким скорее устраняют дефицит витаминов, а не действуют как лекарства. Интересно, что mito-Q, созданный путем прикрепления липофильной катионной группы к убихинону, может накапливаться в митохондриях и действовать там аналогично витамину Е [240]. Однако отсроченные последствия сверхфизиологического повышения убихинона в митохондриях пока неизвестны.
Достижение эффективных концентраций in vivo
Другой проблемой является то, что соединения, индуцирующие системы антиоксидантной защиты, могут не достигать нужных концентраций in vivo, хотя это можно преодолеть в случае с цианоенонами [194]. В условиях, когда адекватное количество активаторов NRF2 поступает с пищей, их медикаментозная доставка не принесет пользы. Кроме того, при окислительном стрессе NRF2 в определенной степени уже активирован, и потенциал для его дальнейшей активации ограничен. Поскольку у пациентов (у которых мы предполагаем наличие окислительного стресса) в клинических испытаниях предполагается хорошее питание, отсутствие усиления антиоксидантной защиты может быть связано с существующими эффектами индукторов NRF2 в этом самом рационе, и, соответственно, меньшим потенциалом активации NRF2. Возможно, активаторы NRF2 следует рассматривать как аналог витаминам, которых не хватает в рационе у многих людей, а также у пациентов, которым трудно потреблять пищу.
Старение
По мере старения снижается способность электрофилов индуцировать NRF2-зависимую экспрессию антиоксидантных ферментов [241]. Сайленсинг BACH1 нивелирует этот эффект для некоторых NRF2-зависимых генов в первичных человеческих клетках бронхов [242], и, следовательно, ингибирование BACH1 потенциально может стать методом антиоксидантной терапии у пожилых пациентов. Но пожилые пациенты отличаются повышенным риском рака, и активация NRF2 у таких пациентов может оказаться нежелательной. Хотя активация NRF2 долгое время ассоциировалась с хемопрофилактикой [243], неприятной стороной является то, что активация NRF2 также защищает опухолевые клетки от окислительного повреждения и может способствовать росту опухоли [200-202]. Тем не менее, сайленсинг BACH1 у мышей, кажется, не усиливал p53-опосредованный онкогенез [244]. Остается надеяться, что дальнейшие исследования прояснят особенности NRF2-связанного онкогенеза, и будут найдены дополнительные способы повышения антиоксидантной защиты без дополнительных рисков для пожилых пациентов.
Перспективы
Окислительный стресс является составной частью патогенеза многих заболеваний, поэтому разработки терапии на основе антиоксидантов сохраняют свой статус важного направления. Несмотря на то, что попытки применения низкомолекулярных антиоксидантов сильно разочаровали, сейчас есть надежда на осознание того, что в основе их применения лежали заблуждения, которые в дальнейшем можно преодолеть. Хотя целью антиоксидантной защиты является предотвращение образования •OH и ONOO— путем снижения уровня их предшественников H2O2 и O2•-, рост осведомленности о том, что H2O2 также является важнейшим участником сигнальных путей физиологических процессов, приведет к более точным подходам в антиоксидантной терапии. Кроме того, ограничения, подчеркнутые в этом обзоре (случаи, когда окислительный стресс первичен, а когда вторичен; пренебрежимо малый эффект практически всех ловушек свободных радикалов; сложность в достижении эффективных концентраций in vivo; снижение эффективности активации NRF2 по мере старения) должны учитываться в дальнейшем, чтобы устанавливать достижимые цели и избегать лишних разочарований.
Перспективными являются препараты, нейтрализующие O2•- и H2O2 внутри клеток и в матриксе митохондрий. Миметики СОД, СОД-каталазы и ГП, кажется, тоже могут оказаться эффективными: некоторые из них уже проходят клинические испытания. Должный уровень глутатиона (субстрата ГП) можно поддерживать при помощи эфиров глутатиона и N-АЦЦ. С эфирами глутатиона пока нет клинических испытаний, а N-АЦЦ уже используется для лечения некоторых отравлений и заболеваний. Кроме миметиков антиоксидантных ферментов и глутатиона, к другой важной стратегии относится повышение синтеза антиоксидантных ферментов и глутатиона через сигнальные пути NRF2 [99]. Авторы ожидают, что все эти подходы будут способствовать развитию антиоксидантной терапии и надеются, что этот обзор будет вдохновлять и информировать о рациональных направлениях в этой области.


