

Апластическая анемия (АА) — это заболевание, характеризующееся панцитопенией, гипоклеточностью костного мозга при отсутствии аномальных клеток или фиброза костного мозга. Первое описание апластической анемии было получено в 1888 году Паулем Эрлихом — это была молодая беременная женщина, умершая от тяжелой анемии и нейтропении, на аутопсии было выявлено замещение межтрабекулярных пространств костного мозга жировой тканью, т. е. отсутствие гемопоэза. Термин «апластическая анемия» был введен французским гематологом А. М. Шоффаром в 1904 году и применен стохастически. Хоть АА и не является распространенным заболеванием, трагичность каждого отдельного случая и фатальные последствия вызывают к ней значительный интерес [1, 3, 4].
АА может носить как наследственный, так и приобретенный характер. Несколько редких врожденных заболеваний, включая анемию Фанкони, синдром Швахмана-Даймонда, врожденный дискератоз, первично характеризуются апластическим типом гемопоэза [2].
В большом количестве литературных источников начиная с XX века описаны различные химические и лекарственные соединения, вызывающие это заболевание; широко известно влияние на показатели крови бензола, а также случаи апластической анемии после введения хлорамфеникола. Хлорамфеникол является широко известным лекарством, документально зарегистрированным как фактор развития апластической анемии. Хотя этот препарат при очень высокой дозе оказывает непосредственно миелосупрессивное действие из-за его влияния на митохондриальную ДНК, возникновение апластической анемии носит идиосинкразический характер, предположительно связанный с унаследованной чувствительностью к нитрозосодержащим токсичным промежуточным продуктам. Риск развития апластической анемии у пациентов, получавших хлорамфеникол, составляет примерно 1 на 20 000, то есть в 25 раз больше, чем в общей популяции. Хотя в индустриальных странах его использование в качестве антибиотика прекращено, до сих пор появляются сообщения о фатальной апластической анемии при его топическом или системном использовании.
Известно, что некоторые препараты также могут вызывать селективные цитопении и агранулоцитоз, которые обычно обратимы и проходят после прекращения воздействия агента. Эти обратимые реакции не коррелируют с риском апластической анемии, ставя под сомнение эффективность рутинного мониторинга клинического анализа крови в качестве метода диагностики апластической анемии [1].
Исследования in vitro и данные клинических наблюдений привели к выводу, что основой для большинства случаев приобретенной апластической анемии служит атака цитотоксическими Т-лимфоцитами CD34+ клеток и стволовых клеток крови (СКК). Иммунное повреждение клеток костного мозга после лекарственной, вирусной или токсической аплазии костного мозга может быть результатом индукции неоантигенов, провоцирующих вторичную Т-опосредованную атаку на кроветворные клетки. Спонтанное или митоген-индуцированное увеличение продукции мононуклеарами интерферона-γ, IL-2 и фактора некроза опухолей-α (TNF-α) ингибирует дифференцировку гемопоэтических клеток [1]. Секреция интерферона-γ является результатом повышения регуляции транскрипционного фактора T-bet и апоптоза CD34+ клеток, частично опосредованного FAS-зависимым путем [3].
В ранних лабораторных экспериментах удаление лимфоцитов из апластического костного мозга увеличивало число колоний клеток в культуре тканей, а их внедрение в нормальный костный мозг подавляло гематопоэз in vitro. Эффекторные клетки были идентифицированы при помощи иммунофенотипирования активированных цитотоксических CD8+ Т-клеток, экспрессирующих Th1-цитокины, в частности γ-интерферон. CD8 клетки могут определяться непосредственно в крови при помощи:
- проточной цитометрии T-клеточного рецептора (TCR);
- спектрального анализа комплемент-определяющего региона (CDR2);
- секвенирование области CDR3 для установления молекулярного клона.
Также снижение количества регуляторных Т-клеток (CD4+, CD25+, FoxP3+) способствует увеличению аутореактивной популяции CD8+ CD28– Т-клеток, которая индуцирует апоптоз аутологичных гемопоэтических клеток. Т-регуляторные клетки являются компонентами иммунной системы, подавляющими иммунные реакции других клеток, а также играющими роль в предотвращении аутоиммунных реакций [2].
АА может возникать из-за метаболической или иммунологической предрасположенности (полиморфизма генов) у восприимчивых лиц. В случае фенилбутазон-ассоциированной аплазии костного мозга происходит замедление процессов окисления и клиренса соответствующего соединения, ацетанилида, по сравнению с нормальной контрольной группой, что предполагает избыточное накопление лекарственного средства как потенциального пускового механизма развития аплазии.
Для пациентов как с приобретенной, так и с наследственной апластической анемией (Фанкони, врожденный дискератоз) характерно наличие дефекта теломераз и восстановления теломер. Одной из характерных черт лейкоцитов при апластической анемии является укорочение теломер, причиной чему предполагали деплеции стволовых клеток. Однако анализ наследования в больших родословных продемонстрировал, что X-связанная форма врожденного дискератоза была вызвана мутациями в DKC1 (dyskeratosis congenita 1), идентификация мутаций в TERC (Telomerase RNA Component) у пациентов с аутосомно-доминантным наследованием помогла выявить генетическую основу укорочения теломер [3].
Центральную роль в восстановлении структуры РНК играет шаблон РНК, кодируемый TERC, при этом теломераза, представляющая собой обратную транскриптазу, кодируемую TERT (Telomerase Reverse Transcriptase), удлиняет нуклеотидную последовательность; другие белки, включая дискерин, кодируемый DKC1, связаны с восстановлением теломер. Систематические исследования ДНК выявили мутации TERC и TERT у некоторых пациентов с приобретенной апластической анемией. У членов семей, имеющих данную мутацию, несмотря на нормальные или почти нормальные показатели крови, выявили уменьшенное количество CD34-клеток, плохую скорость образования гемопоэтических колоний, повышенный уровень гемопоэтического фактора роста, гипоцеллюлярные участки в костном мозге, и, конечно, короткие теломеры. Клиническое проявление этих аномалий проявлялось позже, чем при типичном дискератозе, и не вызывало характерных физических отклонений. Некоторые из пациентов с синдромом Швахмана-Даймонда (Shwachman-Bodian-Diamond) также имеют мутации гена SBDS.
АА может сосуществовать или, по-видимому, эволюционировать вместе с другими гематологическими заболеваниями, характеризующимися пролиферацией специфических клонов клеток, такими как пароксизмальная ночная гемоглобинурия (ПНГ) или миелодиспластический синдром (МДС). Наличие небольшого количества клонов также создает проблемы при постановке диагноза апластической анемии даже при использовании таких высокочувствительных методов диагностики, как фенотипический (проточная цитометрия для ПНГ) или цитогенетический (флуоресцентная гибридизация in situ (FISH) для MDS) анализ [1, 2].
Более пятидесяти процентов пациентов с АА имеют клон ПНГ-клеток, которые обнаруживаются проточной цитометрией по отсутствию мембранных белков, связанных с якорным гликозилфосфатидилинозитолом. Мутация в гене PIG-A в стволовой клетке нарушает синтез гликозилфосфоинозитола (GPI) и, следовательно, вызывает отсутствие экспрессии GPI-связанных белков на поверхности эритроцитов потомства, лейкоцитов и тромбоцитов (CD14, CD16 и CD24 для лейкоцитов, CD55 и CD59 для эритроцитов). Большинство колоний клонов этих клеток небольшие и не приводят к клиническим проявлениям гемолиза или тромбоза, но при ПНГ может преобладать разрушение костного мозга («апластическая анемия/синдром ПНГ»). Было высказано предположение о том, что при ПНГ значительное уменьшение количества белков на клеточной поверхности позволяет «избежать» атаки и выжить существующему мутантному клону. Ассоциация клона ПНГ с HLA-DR29, по-видимому, также позволяет им избежать иммунной атаки и служит предиктором отсутствия реакции на иммуносупрессивную терапию. Тем не менее существует мало конкретных экспериментальных доказательств различия в дифференцировке типов иммунных реакций или восприимчивости клонов ПНГ по сравнению с фенотипически нормальными популяциями клеток-мишеней.
Конечным результатом иммуноопосредованного повреждения костного мозга является уменьшение образования клеток крови в костном мозге. Количество CD34+ клеток и их производных у пациентов с апластической анемией заметно снижено [2].
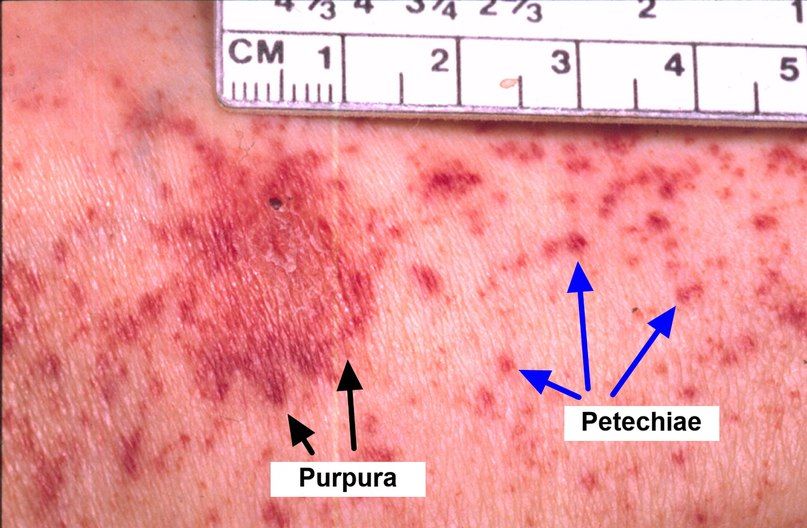
Клинически AA не сопровождается увеличением лимфатических узлов, печени или селезенки. Основным проявлением заболевания является панцитопения: для клинического анализа крови характерно уменьшение содержания всех форменных элементов. На ранних стадиях можно наблюдать изолированную цитопению, чаще тромбоцитопению. Может присутствовать моноцитопения, что требует дифференциального диагноза с волосатоклеточным лейкозом. АА сопровождается снижением ретикулоцитарного индекса, относительное число ретикулоцитов обычно меньше 1 % и может быть равно нулю, абсолютное число ретикулоцитов — менее 40 000 в мкл (40×109/л), несмотря на высокие уровни эритропоэтина; анизоцитоз и пойкилоцитоз отсутствуют. Эти изменения в периферической крови сопровождаются гипоклеточностью костного мозга без аномальных или злокачественных клеток или фиброза. Необходимо тщательное исследование мазков крови для исключения наличия диспластических клеток. При АА может быть увеличено содержание фетального Hb: у детей это требует проведения дифференциального диагноза с миелопролиферативными миелодиспластическими синдромами, такими как ювенильный миеломоноцитарный лейкоз или другие подтипы МДС [3, 4].
Критерии диагностики АА:
- концентрация гемоглобина (Hb) < 100 г/л;
- количество тромбоцитов < 50 × 109/л;
- количество нейтрофилов < 1–5×109/л;
- содержание ретикулоцитов < 60 × 109/л;
- фрагменты костного мозга в аспирате — гипоцеллюлярные с жировым замещением;
- отсутствие диспластических мегакариоцитов и бластных форм; их присутствие указывает на гипопластическую МДС либо эволюцию лейкемии

АА подразделяется на:
- Нетяжелую: отсутствие признаков тяжелой АА [4].
- Тяжелую: клеточность костного мозга < 25 % (или 25–50 % с < 30 % остаточных гемопоэтических клеток) + по меньшей мере 2 критерия из 3:
— количество нейтрофилов < 0–5×109/л;
— число тромбоцитов < 20×109/л;
— количество ретикулоцитов < 20×109/л. - Очень тяжелую: те же признаки, что и при тяжелой, но количество нейтрофилов < 0–2×109/л;
Основным методом диагностики является проведение трепанобиопсии. Аспират костного мозга при АА обычно содержит трабекулы с пустыми, заполненными жиром межтрабекулярными пространствами и малым количеством гемопоэтических клеток. Могут присутствовать единичные лимфоциты, плазматические клетки, макрофаги и тучные клетки [1].
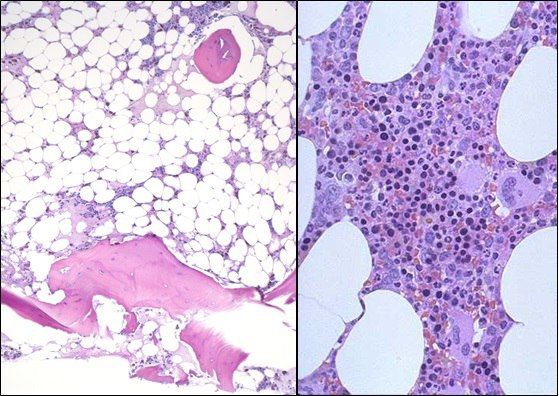
Слева — апластическая анемия, справа — норма
При выполнении цитогенетического анализа могут возникнуть трудности из-за низкой клеточности материала: для получения достаточного количества клеток может потребоваться несколько аспираций. Обнаружение клональных цитогенетических аномалий при апластической анемии является признаком наличия миелоидного заболевания. Переход к более новым методам, таким как сравнительная геномная гибридизация (CGH), позволяет обнаруживать анеуплоидии, делеции, дублирование и/или амплификации любого локуса.
Лучшим доказательством ключевой роли иммуноопосредованного механизма в патофизиологии АА является положительный ответ на иммуносупрессивную терапию у больных с апластической анемией: большинство пациентов демонстрируют гематологическое улучшение после транзиторного разрушения Т-клеток антитимоцитарным глобулином (ATGs), в случае рецидива ответ на ATG также сохраняется. По мере усиления иммуносупрессивной терапии, начиная с ранних попыток лечения при помощи кортикостероидов до использования агрессивных препаратов, таких как циклофосфамид в высоких дозах, увеличилось число сообщений о благоприятных результатах лечения, и иммунный механизм стал рассматриваться как ведущий. Влияние различных генетических факторов, микроокружения и индивидуальных характеристик иммунного статуса больного, вероятно, объясняет вариабельность клинической картины и реакций на проводимое лечение [1].
Терапия проводится при помощи следующих препаратов и методов:
- Антитимоцитарный глобулин (antithymocyte globulin, ATG), получаемый путем иммунизации животных (лошади или кролика) человеческими лимфоцитами; при монотерапии ATG клиническое улучшение наблюдается в 50–60 % случаев. Обычно для улучшения результата его назначают в комбинации с циклоспорином. Также на короткий срок назначаются кортикостероиды для снижения вероятности аллергической реакции и развития сывороточной болезни (лихорадка, сыпь и суставные боли), что может произойти приблизительно через 7 дней после введения ATG.
Количество тромбоцитов должно поддерживаться на уровне выше 10×10^9/л (по возможности — выше 20–30×109/л). При отсутствии ответа на ATG через 4 месяца может быть начат второй курс того же или другого препарата. В целом на комбинацию ATG и циклоспорина положительно реагирует до 80 % пациентов; - Циклоспорин показывает высокую эффективность в сочетании с ALG, но у пожилых людей иногда может использоваться и в виде монотерапии;
- Алемтузумаб (антитело против антигена CD52 лимфоцитов) показал эффективность примерно у 50 % пациентов (в небольших исследованиях), и обычно используется только при неэффективности ATG;
- Элтромбопаг (Eltrombopag) стимулирует образование тромбоцитов, а также может привести к увеличению количества эритроцитов и нейтрофилов;
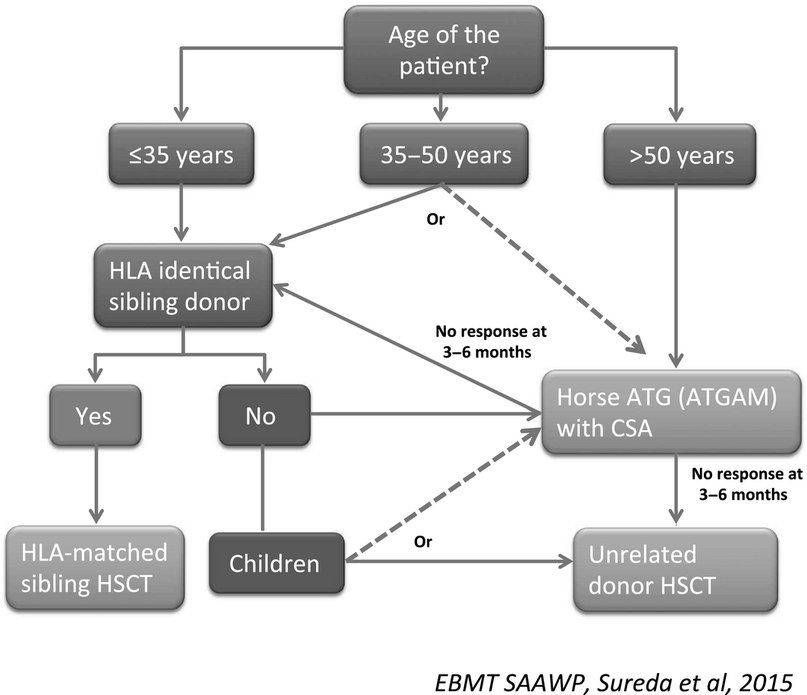
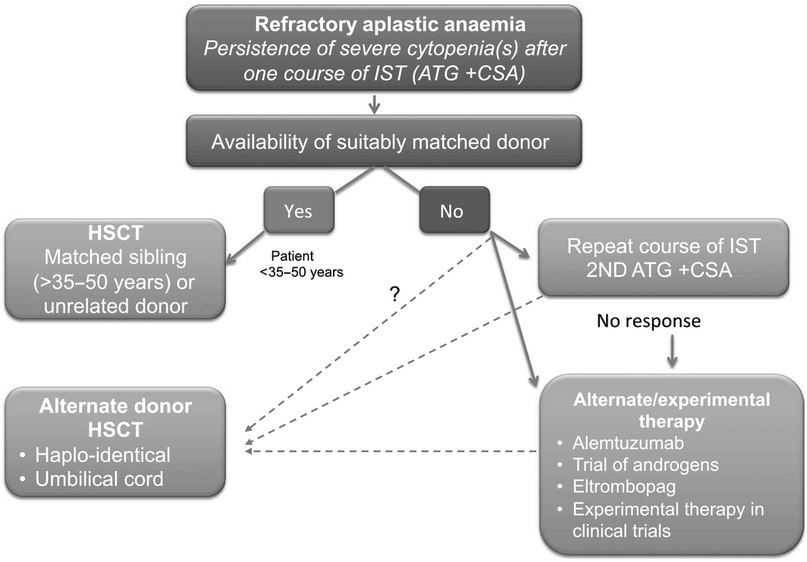
- Трансплантация стволовых клеток (ТСК). Проведение аллогенной ТСК у отдельных пациентов подразумевает возможность полного выздоровления. Кондиционирование проводится с циклофосфамидом без облучения, для снижения риска неприживления трансплантата и реакции «трансплантат против хозяина» используется циклоспорин. Сравнительный анализ эффектов ТСК и иммуносупрессивной терапии у лиц с апластической анемией до сих пор остается предметом дискуссий. Как правило, ТСК применяется у пациентов с тяжелой апластической анемией в возрасте менее 35 лет и имеющимся HLA-совместимым донором. У отдельных пациентов в возрасте старше 40 лет возможно применение ТСК с использованием немиелоаблативных режимов, но в основном у пациентов старшего возраста и лиц с меньшей тяжестью заболевания в первую очередь проводится иммуносупрессия;
- Гемопоэтические факторы роста. Гранулоцитарный колониестимулирующий фактор (G-CSF) может вызывать незначительный ответ, но обычно не приводит к стойкому улучшению. Другие факторы роста не показали свою эффективность [2, 3, 4].
- Иммуносупрессивная терапия 1 линии — это сочетание лошадиного ATG и циклоспорина для пациентов с нетяжелой АА, тяжелых или очень тяжелых пациентов с АА, у которых отсутствует подходящий родственный донор, а также тяжелых или очень тяжелых пациентов с АА старше 35–50 лет. Второй курс ATG может быть проведен после отсутствия ответа на первый курс или после рецидива после первого курса. При отсутствии ответа на второй курс АА считается рефрактерной и проводится альтернативная терапия при помощи алемтузумаба, элтромбопага или ТСК.
Также необходима поддерживающая терапия при помощи гемотрансфузии. Единого целевого значения концентрации гемоглобина не существует, и оно должно быть установлен индивидуально. Для снижения риска иммунизации следует учитывать фенотип Rh и Kell. Пациентам с АА, получающим активное лечение и находящимся в стабильном состоянии, следует назначать профилактические переливания тромбоцитов, пороговая концентрация тромбоцитов составляет 10×109/л. У пациентов, имеющих факторы риска кровотечения, число тромбоцитов должно составлять 20×109/л. Регулярные профилактические переливания тромбоцитов не рекомендуются для стабильных пациентов с АА, не находящихся на активном лечении в данный момент. У пациентов с апластической анемией при регулярной поддержке переливания РБК будет развиваться перегрузка тканевого железа. Сывороточный ферритин остается наиболее широко используемым показателем для оценки перегрузки железом. Магнитно-резонансная томография может определять содержание железа количественно и является хорошим дополнением к лабораторным методам мониторинга [4].


