

Введение
Нобелевская премия 2024 года в естественных науках, повторяя традиции предыдущих лет, оказалась достаточно непредсказуемой. Интересный выбор в области физиологии и медицины, снова показывающий большую важность исследований РНК; неожиданное решение о присуждении премии по физике за разработку основ машинного обучения. Наконец, прекрасная премия по химии выдана Дэвиду Бейкеру за вычислительный дизайн белков, а также Демису Хассабису и Джону Джамперу за предсказание их структур (Рис. 1).

Рисунок 1 — Презентационная картинка Нобелевского комитета о вручении премии по химии в 2024 году.
На просторах сети можно встретить множество критики и обсуждений, связанных с присуждением премии по химии за методы моделирования, в том числе основанные на применении методов глубокого обучения. В действительности, премия 2024 года не является первой в данной области.
Предпосылки
Скорее всего, первым Нобелевским лауреатом, разработавшим метод моделирования, можно считать Роберта Малликена, который в 1966 году получил премию за разработку метода молекулярных орбиталей, позволившего описывать поведение электронов и проводить предсказания в молекулярных системах, состоящих из более чем двух атомов.
В 1998 году Уолтер Кон и Джон Попл получили премию за разработку принципов и методов квантовой химии, позволивших с помощью компьютерных расчетов описывать и предсказывать поведение молекул и систем.
В 2013 году за создание многоуровневых моделей сложных химических систем премию получили Мартин Карплус, Майкл Левитт и Арье Уоршел. Исследователи подружили классическую быструю ньютоновскую механику и чрезвычайно вычислительно дорогую квантовую механику для описания больших систем, это дало неслыханную ранее возможность производить расчеты сложных ферментативных реакций.
И вот в 2024 году снова моделирование. Но что за проблему решали на этот раз?
Проблема фолдинга белка
Еще в 1955 году Фредерик Сэнгер доказал, что белки состоят из аминокислот, соединенных в одиночные цепочки за счет пептидных связей. Последовательность аминокислот в белке называется первичной структурой.
Немногим ранее в 1951 году Лайнус Полинг и Роберт Кори предложили две важные модели вторичной структуры белков, а именно α-спираль и β-лист. В 1958 году Джон Кендрю и Макс Перуц полностью расшифровали пространственные структуры миоглобина и гемоглобина, экспериментально подтвердив модели Полинга и Кори.
Однако было понятно, что в природе белки сворачиваются в сложные пространственные структуры (третичные), за счет которых и выполняют свою функцию. Этот процесс называется фолдингом. В 1961 году Кристиан Анфинсен доказал, что белок принимает свою пространственную структуру за счет информации о первичной последовательности. Бычья панкреатическая рибонуклеаза, фермент, который использовался в исследовании, восстанавливал свою структуру после денатурации. Позднее были обнаружены белки, которым необходима помощь других белков в упаковке — так были открыты шапероны. А затем было показано существование неструктурированных белков (intrinsically unfolded proteins, IDPs), которые принимают несколько разных структур с равной вероятностью, но эти находки не нарушали идеи о связи первичной и третичной структуры. В своей статье 1973 года Кристиан Анфинсен писал: «Соображения о … корреляции между последовательностью и трехмерной структурой … начинают делать более реалистичной идею априорного предсказания конформации белка». С этого момента и можно вести отсчет так называемой «проблемы фолдинга белка» [10.1126/science.181.4096.223]. В 2005 году журнал Science опубликовал список из более чем ста крупнейших проблем современной науки, в котором, наряду с вопросами о природе гравитации и возможности существования сверхпроводников при комнатной температуре, указана и проблема белкового фолдинга [10.1126/science.309.5731.78b].
Неверным было бы предположить, что с 1973 года не было никакой возможности предсказывать третичные структуры белков. В действительности подобные методы разрабатывались постоянно. Их можно условно разделить на две группы:
1) ab initio («из первых принципов») — имея в арсенале огромные знания о физике сворачивания биополимеров, можно попробовать симулировать процесс фолдинга. Симулируя условия, требуемые для сворачивания, а также набор правил поведения молекул, можно произвести длительный расчет, позволяющий имитировать естественные процессы на компьютере. В действительности из-за размеров систем приходится прибегать к массе упрощений для проведения вычислений за адекватное время, в связи с чем падает и точность. Опустив интересную, но долгую историю изысканий, можно прийти к пику карьеры данной группы методов. В 2007 году группа под предводительством одного из героев статьи, Дэвида Бейкера, взялись за определение структуры белка с неизвестной функцией из бактерии Bacillus halodurans, предсказанной из анализа ее генома и состоящей из 112 аминокислот. Предсказав структуру 5 раз с помощью программы Rosetta (разработанной той же группой), исследователи установили участки, значительно отличающиеся между запусками расчетов. Их было принято считать ошибочными и перестроить отдельно. Для этого данные участки вырезали и исследовали широкое разнообразие их возможных конформаций, после чего выбирали наиболее энергетически выгодное состояние, и вставляли на место. Данный подход в 1 из 26 случаев позволил построить систему, невероятно схожую с экспериментально определенной структурой. Еще в 7 результатах конформации были не совсем правильными, но могли быть полезны в экспериментальной расшифровке структур. Для всего исследования были использованы мощности более 70 тысяч персональных компьютеров добровольцев из проекта Rosetta@Home, а также суперкомпьютер IBM Blue Gene/L.
Одной из очень неприятных проблем, связанных с предсказанием на основе ab initio методов, являются шапероны. Шапероны — это особый класс белков, которые играют ключевую роль в правильном сворачивании других белков. Некоторые из них не могут принимать правильную форму без шаперонов, из-за чего могут агрегировать внутри клетки и полностью терять свою функцию. И очевидно, что моделировать процесс взаимодействия данного белка с шапероном при ab initio построении чрезвычайно сложно, если не невозможно на текущем этапе.
Результат исследования лаборатории Бейкера 2007 года хоть и прорывной для отрасли в целом, но вряд ли способен впечатлить людей, не знающих таящихся за ним сложностей. Но не стоит хоронить ab initio методы совсем, отражение их идей мы еще обсудим позже.
2) Построение по гомологии — обратившись к основополагающим идеям биологии, а именно связи структуры с функцией, а также теории возникновения новых белков из других, мы сразу догадываемся, что, зная структуру некоторого количества белков, мы, фактически, способны достроить всех их родственников. Другими словами, если первичная структура (практически) однозначно сворачивается в какую-то третичную, то одинаковый или очень близкий по последовательности ее участок в двух разных белках должен сворачиваться единообразно. В принципе, в этом и заключается построение по гомологии. Мы можем произвести поиск последовательностей (например, с помощью алгоритма BLAST) гомологичных нашему белку среди структур, пространственная форма которых известна. Далее, используя эту информацию, мы можем построить структуру интересующего нас белка. Данная тривиальная идея большую часть 50-летней истории проблемы фолдинга белка являлась основным способом моделирования структур неизвестных белков. Ограничение данной группы методов также довольно тривиально: если у какого-то участка белка нет гомологов с разрешенной структурой, мы не можем его так построить, но с другой стороны, с этим уже можно работать.
Конечно, это не единственные подходы, но наиболее качественные результаты обычно выдавал подход построения по гомологии, иногда с достраиванием неизвестных участков с помощью ab initio методов.
И восстали машины из пепла…
Прогресс в предсказании структур белков пусть и сохранялся, но развивался с не очень привлекательной динамикой. Для отслеживания успехов в решении данной проблемы в 1994 году было учреждено соревнование CASP (Critical Assessment of techniques for protein Structure Prediction), которое на неопубликованных расшифрованных структурах белков проводило честное сравнение методов предсказания, предложенных разными разработчиками.
Если в 1994 году методы предсказания позволили правильно установить только около 15–26 % положений аминокислотных остатков в сложных белках, то к 2014 этот процент едва дотягивал до 35. И вот, наконец, в 2018 году появляется совершенно новый подход — AlphaFold.
Но давайте сначала углубимся в алгоритмы, стоящие за нашумевшей революцией.
Искусственные нейронные сети
Не окунаясь в историю искусственных нейронных сетей, мы попытаемся в упрощенной форме понять, что это за сущности.
Итак, начнем с простого:
Допустим, у нас есть некоторая математическая функция, которая описывает известное нам явление. Скажем, пускай это будет размножение кроликов в условиях заповедника. Легко догадаться, что эта функция чем-то похожа на экспоненту (на самом деле это не так, но это безумие выходит за рамки статьи), однако предположим, что мы этого не знаем. Вот мы как-то хотим вывести эту функцию из наблюдений за кроликами. К счастью, математика дает нам такую возможность, благодаря интерполяционному многочлену Лагранжа. Сразу должен отметить, что это всего лишь способ сделать некоторую наиболее простую в смысле используемых степеней функцию, которая попадает во все наблюдаемые нами точки. В нашем примере с кроликами мы будем добавлять в некоторую таблицу значения количества кроликов в каждый день, неделю или месяц наблюдений, после чего применим какую-то математическую магию и получим функцию, которая предсказывает численность кроликов в промежуточных этапах и, возможно, в будущем. Данная функция не является самым простым и точным описанием наблюдаемого явления, а лишь аппроксимирует (то есть приближенно отражает) это явление, поэтому и называется аппроксимирующей (Рис 2).
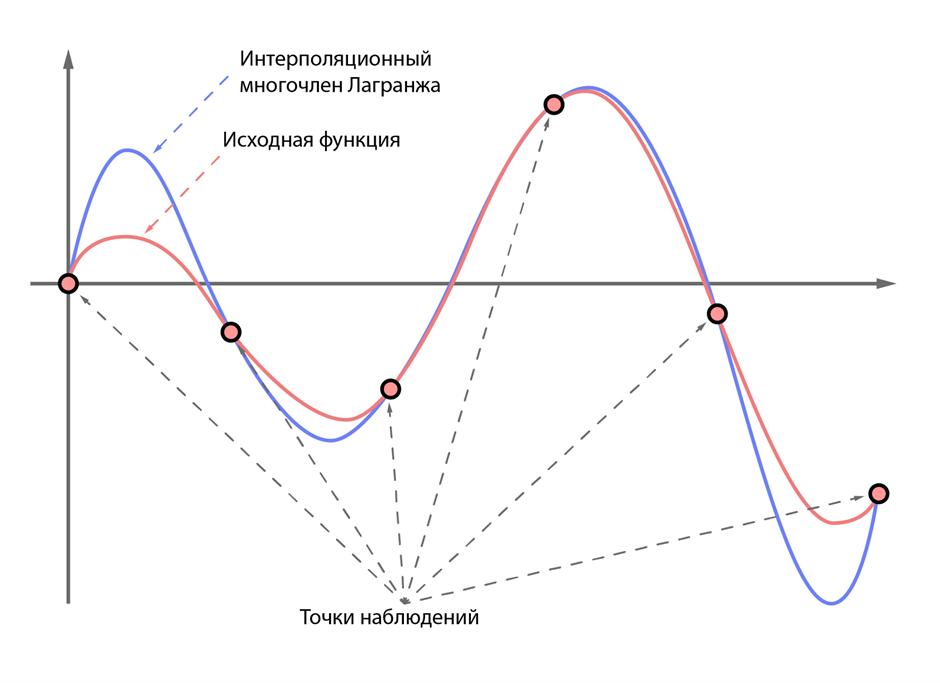
Рисунок 2 — пример интерполяционного многочлена Лагранжа для некоторой сложной функции.
А что, если мы не можем определить какое-то конкретное значение, которое численно можно записать? Математика нам намекает, что любую закономерность можно описать некоторой функцией. Но не всегда эта задача проста. Если популяцию кроликов мы можем условно представить как зависимость количества кроликов от времени (двухмерное пространство), то как себе представить зависимость фраз на английском языке от их перевода на русском? Данная закономерность явно не является простой, например двухмерной. В действительности для математиков не является проблемой навешивать любое количество размерностей для достижения цели, однако искать аппроксимирующую функцию в огромном количестве пространств крайне сложно. Здесь как раз в игру и вступают нейронные сети. Если описать идею нейронной сети двумя словами, то получится «универсальный аппроксиматор». Подобрав достаточное количество искусственных нейронов, являющихся по сути членами некоторой степени в том самом многочлене Лагранжа с дополнительными множителями перед ними, которые отражают их значимость (более известную как «вес»), мы можем «подогнать» любую функцию по наблюдаемым явлениям под предсказание промежуточных или близких вещей. Если какой-то нейрон не имеет значения и его вес равен нулю, то умножение любого значения данного нейрона на ноль не изменит результат. То самое «обучение», о котором много говорят последнее время, есть ни что иное, как подбор весов отдельных нейронов в процессе «подгонки» функции к наблюдаемым явлениям.
Что ж, звучит не так уж и страшно. Но теперь давайте подумаем, а как бы эту задачу прикрутить к чему-то сложному? Например, к переводу человеческой речи с одного языка на другой. Очевидно, размерность той самой функции здесь будет чудовищной. Мы все знаем, как одни и те же слова могут означать абсолютно разные вещи в разных контекстах. Скажем, «basic solution» может переводиться как «основный раствор» если вы химик, но в тоже время в другом контексте это может означать «базовое решение». И как с этим жить?
Attention!
В 2017 году Ашиш Васвани, Ноам Шазир, Илья Полосухин и другие авторы опубликовали в базе препринтов arXiv статью под названием «Attention Is All You Need», где подробно разобрали архитектуру разработанной ими модели трансформера для обработки информации, требующей понимания последовательности данных. Самой главной частью модели трансформера является механизм внимания (attention). Представим, что нам нужно читать некоторое очень большое предложение. Мы можем читать его слово за словом, но тогда в какой-то момент мы начнем забывать, с чего оно начиналось. С другой же стороны, мы можем посмотреть на общий контекст слов в предложении, а затем связать их между собой, что будет гораздо более простой задачей. Более того, мы без труда можем понять, какое слово имеет важное значение, а какое менее важно для данного предложения. Вернемся к модели трансформера. Итак, в процессе обработки большого количества текстов на разных комбинациях слов она обучается оценивать, какие слова имеют большее или меньшее значение. При этом посмотреть на важность разных слов можно «под разным углом», поэтому вместо одного такого механизма внимания можно добавлять несколько, что называется «многоголовый механизм внимания» или «multi-head attention». Удивительным образом этот простой механизм оказался чрезвычайно эффективным. Именно за счет трансформеров и появилось такое разнообразие дипломных работ (зачеркнуто) моделей, вроде GPT (Generative Pre-trained Transformer), Grok, Gemini и тд.
Обучив механизм внимания находить скрытые закономерности в словах, мы получили сложную функцию, описывающую естественный язык. Ее размерность невероятно большая. Если говорить грубо, то количество пространств этой функции равно количеству семантических значений слов во всех текстах, которые прошли через модель в процессе обучения. Ярким примером того, что закономерности были действительно изучены, является простое наблюдение. Если из всего семантического пространства мы вытащим такие слова как кот, кошка, мужчина и женщина, то заметим, что расстояния между мужчиной и женщиной, а также котом и кошкой будут одинаковыми, точно так же будут равными попарные расстояния между самцами и самками данных видов (Рис. 3). Более того, если мы попытаемся выяснить, как называется женщина кот, то нам будет достаточно добавить направление и расстояние между мужчиной и женщиной к коту, и мы точно попадем в кошку. Фантастика.
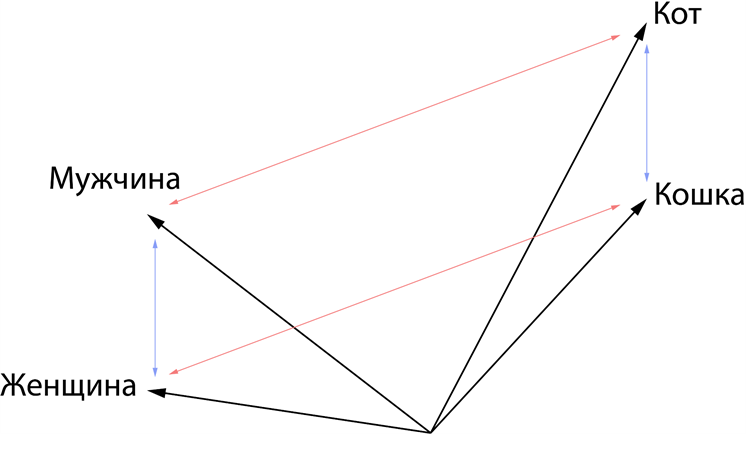
Рисунок 3 — результат обучения механизма внимания, приводящий к сохранению попарных расстояний между различными семантиками.
AlphaFold
Но если последовательностью можно считать любой набор символов, чем белки хуже? Собственно, Демис Хассабис и Джон Джампер подумали так же. В 2018 году они применили нейронную сеть к анализу взаимосвязей между аминокислотами в известных структурах белков. Вместо того чтобы предсказывать положения отдельных атомов аминокислот в белках, они предположили, что более эффективно будет использовать информацию о взаимных углах и расстояниях между аминокислотами в белках. В дополнение использовалась информация о множественном выравнивании последовательности искомого белка и всех известных структур белков на данный момент. И это сработало. В 2018 году AlphaFold внезапно показал медианную точность предсказания структуры на уровне около 58 %. В следующей версии, AlphaFold2, Хассабис и Джампер добавили анализ взаимосвязей между аминокислотами за счет модели трансформера. Модель уже запомнила все известные на тот момент белки и строить множественное выравнивание было не нужно. Более того, модель имела возможность многократно корректировать промежуточные состояния в процессе предсказания. И в 2020 году AlphaFold2 уже набрала 87 % медианной точности предсказания, а это, на секунду, сопоставимо по точности с экспериментальными методами расшифровки (Рис. 4).
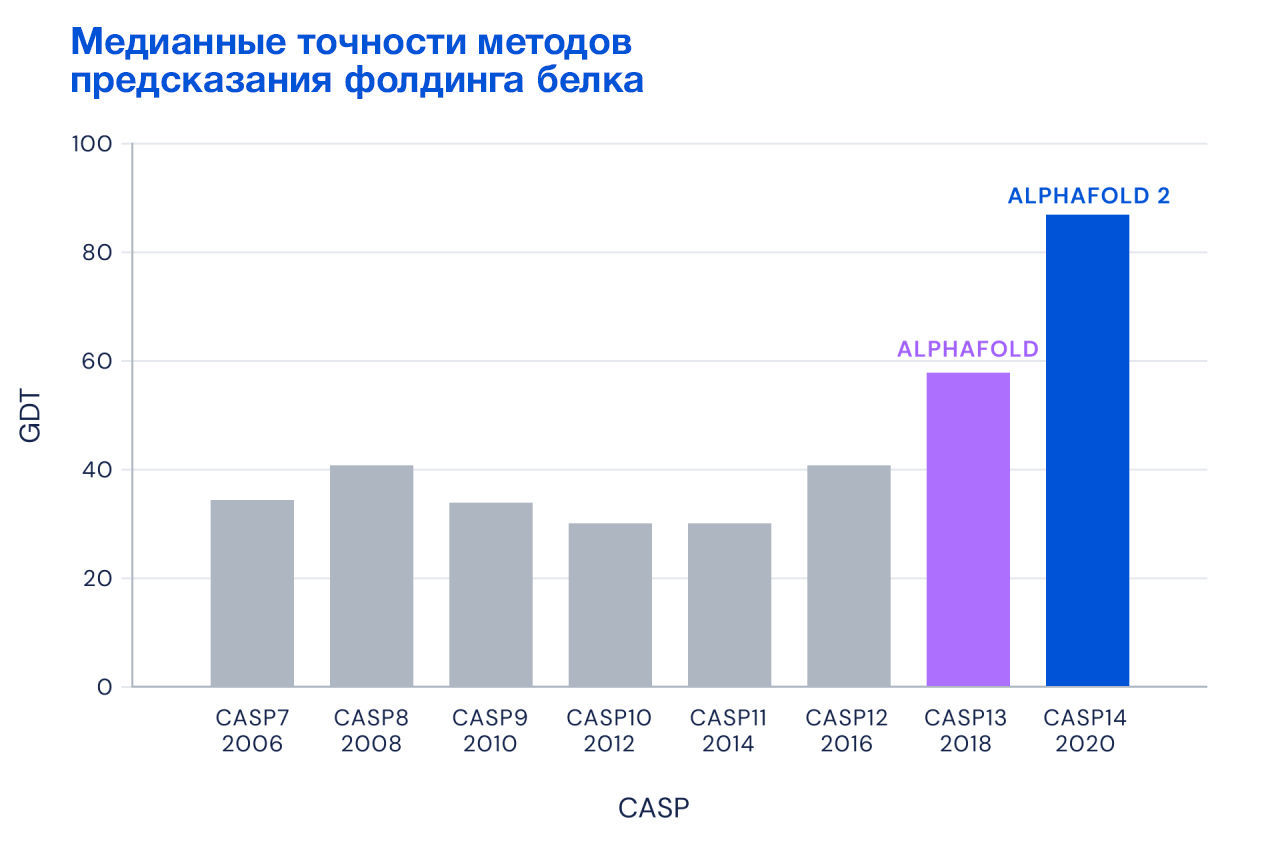
Рисунок 4 — сравнение медианных точностей методов предсказания фолдинга белка в соревнованиях CASP с 2006 по 2020 годы (источник: [https://deepmind.google/discover/blog/alphafold-a-solution-to-a-50-year-old-grand-challenge-in-biology/])
Прогресс был настолько значимым, что такие базы данных как UniProt и PDB добавили структуры, предсказанные AlphaFold2 (и хранящиеся в AlphaFold Protein Structure Database), в дополнение к экспериментальным структурам.
AlphaFold2 оказался настоящим прорывом в решении проблемы фолдинга белков. Но в чем в действительности заключается его принцип работы?
Беря за основу информацию об известных структурах белков и находя сходства с ними, мы строим структуры исследуемых белков с помощью нейронной сети. Несложно догадаться, что данный подход является «нейронным» развитием метода построения по гомологии. И с ним не все так замечательно.
Большинство известных структур белков получены путем рентгеновской кристаллографии. Часть этих структур очень сложно расшифровать, например, потому, что они находятся внутри мембраны клетки, или слишком подвижные. И такие элементы обычно вырезаются из структур, загруженных в базу данных. К сожалению, из-за этого AlphaFold, который обучается на этих данных, понятия о таких структурах не имеет. Поэтому довольно часто AlphaFold выдает что-то невнятное, честно признаваясь «здесь мои полномочия все» (Рис 5). Нередки бывают случаи, когда структура гомолога представляла собой комплекс белка с другой молекулой, но AlphaFold игнорирует эту информацию и строит белок вне комплекса с точно такой же конформацией [10.1016/j.sbi.2022.102526].
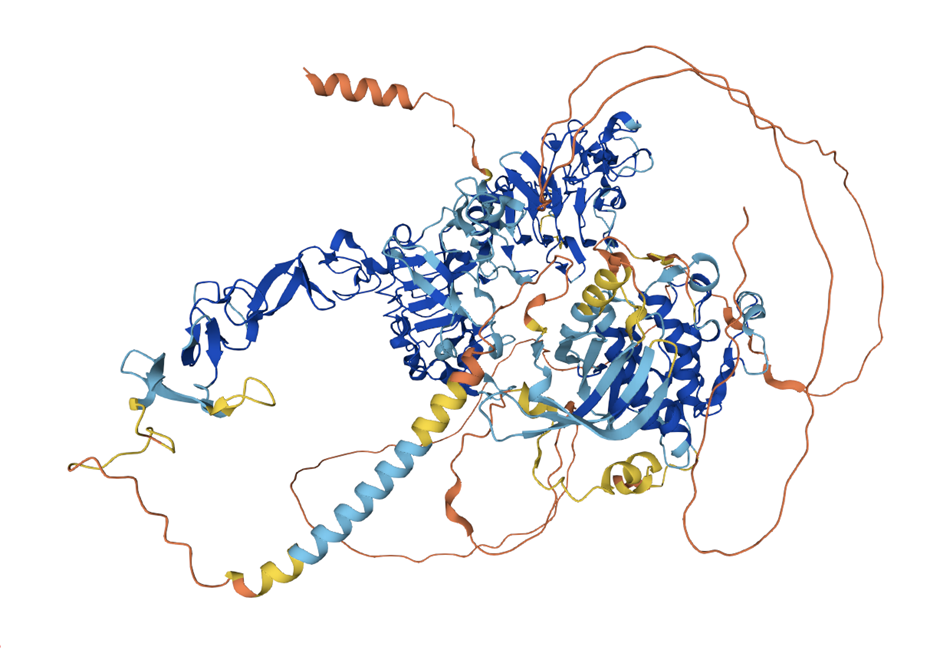
Рисунок 5 — пример предсказания AlphaFold2. Четко видны области, для которых структуры были экспериментально разрешены ранее (синие), и области, у которых не было известных гомологов в обучающей выборке (оранжевые).
Однако в целом эти недостатки не так существенны, поэтому на основе полученных с помощью AlphaFold данных можно исследовать функции некоторых малоизученных или неизвестных белков, разрабатывать потенциальные лекарства и многое другое.
Что там с ab initio?
Существует необычная проблема нейронных сетей, когда им увеличивают «глубину». Если к одной группе нейронов подключить вторую, а затем третью и т.д., то с одной стороны это приведет к более сложным нелинейным зависимостям и улучшению возможностей нейронной сети, а с другой — потребует большего количества данных для ее обучения. Связан последний факт с так называемым затухающим градиентом. Проходя от слоя к слою, информация, которая используется для подбора весов, становится все менее значимой, и изменения становятся более слабыми. Чтобы решить эту проблему в 2015 году группа исследователей из Microsoft Research разработали модель ResNet, в которой информация может «перескакивать» через слой или несколько слоев для усиления.
Данную модель использовали в 2019 году для обучения нейронной сети предсказывать взаимные расстояния между атомами в белке. Вместо того, чтобы обучать модель строить белки по гомологии, исследователи разделили белки на группы и обучали модель только на отдельных группах, а тестировали на тех, которые им не гомологичны. Так удалось обучить модель выявлять сложные зависимости взаимных расстояний между атомами в зависимости от первичной структуры, то есть модель буквально научилась «понимать» те самые «первые принципы». Данная модель показала 65,7 % медианной точности предсказания структур белков, для которых неизвестны гомологи.
В целом, опять можно сказать, что комбинация построения по гомологии и ab initio дает наиболее точные результаты, но в чем же революция? А дело все в том, что AlphaFold, не в последнюю очередь благодаря маркетингу, привлек внимание биологов к ранее редко упоминаемому в биологических кругах инструменту — языковым моделям. Так, уже в 2021 году появилась модель EMSFold, которая не использовала вообще никакого выравнивания и из голой аминокислотной последовательности строила модели с примерно той же точностью, что и AlphaFold2. Затем появилась похожая, но более быстрая модель EMBER3D. Сами авторы тоже не остановились на достигнутом и еще позднее в 2023–2024 годах появились AlphaFold3, AlphaProteo и AlphaFold-Multimer, но об этом позже.
Где же Бейкер?
Как истинный философ, Дэвид Бейкер (когда он поступил в Гарвардский университет, то сначала выбрал философию и социальные науки, но затем влюбился в молекулы и посвятил жизнь им) думал в другую сторону. «А что, если по пространственной структуре белка предсказать его последовательность?».
Вопрос неожиданный, но, с другой стороны, пространственная структура определяет функции, и если мы выберем интересную нам функцию, построим подходящую под нее структуру и получим воспроизводящую ее последовательность, то мы буквально сможем сделать полезный искусственный белок.
Стоит уточнить, что на самом деле идея была не новой. Еще в 1992 году в институте белков под авторством таких известных исследователей, как Финкельштейн и Птицын, вышла статья, в которой был спроектирован первый искусственный белок, не выполняющий какой-то интересной функции, но при этом и не встречающийся в природе. Он состоял из двух α-спиралей и одного β-листа, из-за чего получил вполне логичное название альбебетин [10.1016/0022-2836(92)90092-X].
Бейкер вряд ли знал об этом исследовании, но пошел по тому же пути. В 2003 году он с коллегами опубликовал работу, в которой также получил белок, у которого не было «родственников» в природе (Рис. 6). Белок top7 состоял из 97 аминокислот и тоже включал две α-спирали и один β-лист. Предсказание и последующая экспериментальная расшифровка оказались чрезвычайно близкими по своей структуре. Для данного предсказания использовалась программа RosettaFold, разработанная в той же лаборатории.

Рис 6 — структура белка top7, спроектированная с помощью программы RosettaFold в лаборатории Бейкера.
Название Rosetta отсылает к Розеттскому камню, на котором были написаны 3 идентичных текста на древнегреческом и двух разных египетских языках, благодаря чему стало возможным расшифровать египетские иероглифы. Собственно, именно эта мысль была заложена в программу RosettaFold — научиться переводить с языка аминокислот в белке на язык углов и расстояний в нем.
Получив первый дизайнерский белок, команда Бейкера не остановилась, и они замахнулись на новый рубеж — добавить белку функцию. Любые белки, специфически связывающиеся с малыми молекулами (лигандами), содержат определенные сайты для такого связывания. Немного упрощая концепцию, данные сайты можно представить некоторыми ямками в структуре белка, где аминокислоты ориентированы особым образом, чтобы по электростатическим, гидрофобным и геометрическим свойствам совпадать со структурой молекулы-лиганда. Исследователи лаборатории решили сделать следующее — собрать самые подходящие аминокислоты для связывания вокруг молекулы лиганда, а затем подобрать подходящие каркасы белков, чтобы эти аминокислоты объединить. Программа Rosetta была адаптирована под данную задачу и получила название RosettaMatch. Уже в 2008 году команда разработала фермент, способный стабилизировать переходное состояние в определенной химической реакции, за счет чего ускорить ее течение. Хотя в данной работе после проектирования была проведена т.н. in vitro эволюция, основой для нее было компьютерное предсказание структуры [10.1038/nature06879]. В 2017 году команда опубликовала статью, в которой разработала и проверила в лаборатории синтетический белок, способный специфически связывать фентанил. Полученный белок даже можно было использовать в целях определения данного наркотического средства в крови [10.7554/eLife.28909].
Конечно, создавать дизайнерские белки таким путем это в принципе отличное достижение, но Бейкеру и этого было недостаточно. Необходимость проведения экспериментальной доработки или не 100 % вероятность успеха при синтезе его не устраивали. И тогда он решил использовать глубокое обучение. Но его внимание упало не на языковые модели, а на способ генерации изображений или музыки — диффузионные модели.
Давайте снова отвлечемся от биологии и подробнее рассмотрим, что такое эти диффузионные модели. Диффузия — это неравновесный процесс перемещения частиц из области с высокой их концентрацией в область с низкой, что приводит к самопроизвольному выравниванию системы. Другими словами диффузия — это самопроизвольный процесс изменения положения частиц из более упорядоченного состояния в менее упорядоченное. В 2015 году Яша Соль-Дикштейн и соавторы опубликовали малозаметную работу под названием «Глубокое неконтролируемое обучение с использованием неравновесной термодинамики», в которой использовали идею диффузии, обращенной в противоположную сторону, для генерации чего-то упорядоченного [https://proceedings.mlr.press/v37/sohl-dickstein15.html]. Позже в 2019 и 2020 году было опубликовано 2 работы с очень похожим смыслом [10.5555/3454287.3455354][10.48550/arXiv.2006.11239]. Идея вот в чем: если взять какое-то упорядоченное состояние, скажем, порядок пикселей в изображении, и постепенно превращать их в шум, на каждом шаге обучая модель брать зашумленный вариант и превращать в менее зашумленный, то в результате можно получить довольно полезный инструмент. Конечно, сам по себе такой инструмент полезен разве что для снижения шума на видео, фотографиях или при рендеринге в каких-нибудь играх, однако если добавить ко всему этому текстовое описание исходной картинки, а также способ модели это текстовое описание понимать, то получится генератор изображений. А вот тут поподробнее.
Представим, что мы хотим получить изображение, например, котика, играющего на гитаре. Мы даем модели текст «котик играет на гитаре». Не будем вдаваться в подробности, как модель понимает эти слова, это процесс, который похож на описанные выше трансформеры или даже в точности повторяет их. Однако дальше наступает процесс генерации. Сначала обычный алгоритм создает шум, то есть случайный набор пикселей, затем нейронная сеть шаг за шагом начинает удалять этот шум особым образом, чтобы изображение все больше и больше напоминало котика, играющего на гитаре. А как нейронной сети понять, что она сделала именно котика с гитарой? Дело в том, что модель, обученная на огромном объеме изображений, включающем разные объекты, сцены и композиции, может по чуть-чуть удалять шум в направлении нужной картинки. Этот процесс чем-то похож на написание картины маслом, когда создаются большие и общие детали, тени, свет, а затем постепенно уточняются все более и более мелкие. Дополнительным улучшением в последнее время является необязательное использование классификаторов, способных оценивать результат на еще зашумленном варианте, а именно понимать, что там изображено. Так как учатся они в основном на продуктах человеческого творчества с искусственно добавленным шумом, этот процесс позволяет значительно улучшить качество получаемого изображения. В этом, в частности, кроется причина столь быстрого развития таких программ для генерации изображений как Midjourney, StableDiffusion и других.
Пришло время вернуться к белкам. Дэвид Бейкер, безусловно, не прошел мимо этой идеи и в апреле 2023 года опубликовал модель под названием RFDiffusion (Rosetta Fold Diffusion). Та же идея диффузии применялась для получения пространственных структур белков с заданной геометрией, после чего легко можно было восстановить их последовательность. Не нужно было искать подходящие каркасы белков, модель просто шаг за шагом строила их из случайного набора атомов в пространстве [10.1038/s41586-023-06415-8].
Идея заключается в том, что можно задать некоторое определенное положение аминокислот в пространстве, а вокруг насыпать огромную кучу атомов, входящих в состав аминокислот. Затем диффузионная модель постепенно меняет положение шумных атомов таким образом, чтобы восстановить структуру чего-то, что похоже на белок (Рис 7). Дополнительной оценкой занимается другая программа, которая на основании информации из эволюции белков, а также биофизики, вторичных структур и других факторов определяет, является ли полученная структура похожей на правду. Получив конечную структуру, можно с большой долей вероятности предположить, что при синтезе внутри клетки она будет свернута именно в такую форму. Бейкер даже не постеснялся — перевел полученные в ходе моделирования структуры в первичные, а затем предсказал их фолдинг с помощью AlphaFold2. Результат оказался чрезвычайно близким.
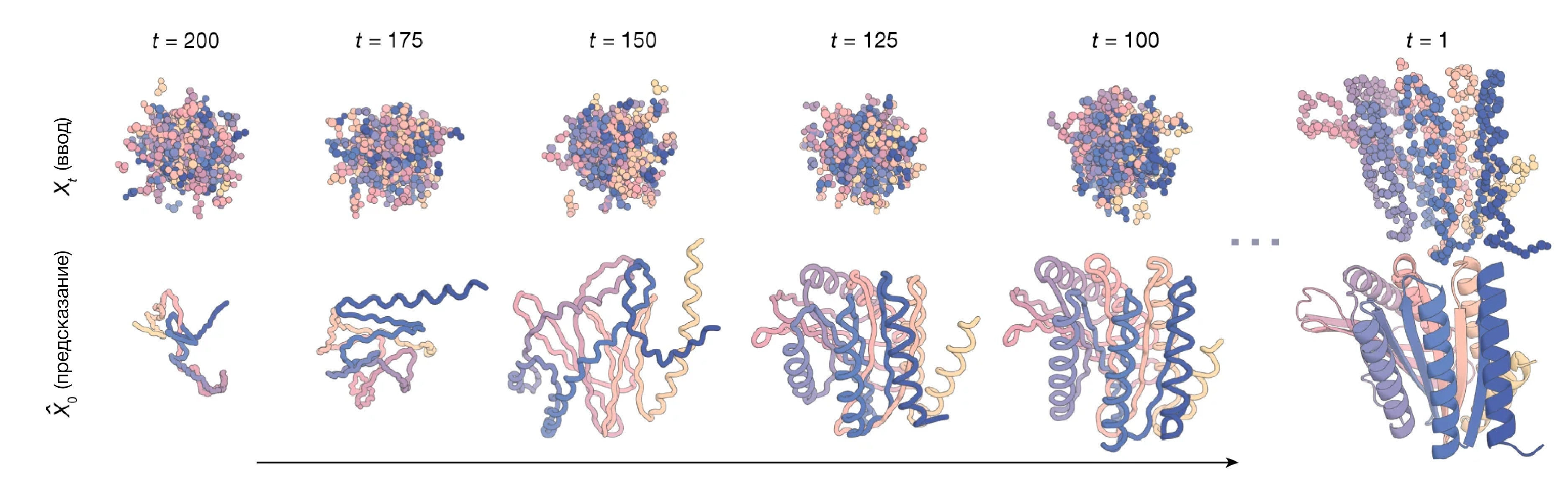
Рисунок 7 — пример построение структуры белка в процессе диффузии RFDiffusion (источник: [10.1038/s41586-023-06415-8]). Сначала получается что-то совсем не похожее на белки, но постепенно структура белка становится все более и более реалистичной.
Не обошлось, правда, и без проблем. Также как AlphaFold, RFDiffusion обучалась в большей степени на данных из PDB, в которой подавляющее большинство структур являются результатами применения рентгеновской кристаллографии. Такие структуры несколько отличаются по форме от нативного состояния, так как подвержены процессу высаливания (кристаллизации белков в упорядоченные, но вместе с тем слегка сжатые структуры). Конечно, данное явление приводит к ошибкам, хотя они вполне неплохо решаются с помощью расслабления в, например, молекулярной динамике. Также сложно использовать данный подход к получению комплексов белков — не учитываются посттрансляционные модификации и многое другое. Вместо применения просто хаотичного набора атомов более надежным является использование заранее подготовленных каркасов, которые уже и дорабатываются моделью, но их подбор требует больших затрат времени и ресурсов.
Тем не менее RFDiffusion позволила сделать ранее небывалые вещи. Одним из ее ярких достижений является доработка природных белков для придания им большей устойчивости при воздействии температур, изменения уровня pH, изменения их растворимости в различных средах и др [10.1021/jacs.3c10941]. Все эти изменения позволяют использовать белки вне живых организмов, например, на производстве, с меньшими трудностями, связанными с их выделением, хранением и подбором условий эксплуатации.
Давайте на секунду задумаемся: диффузионная модель RFDiffusion двигает структуры атомов и проверяет, насколько они совпадают с биофизическими и другими накопленными данными. С небольшой поправкой на направленность данного процесса несложно догадаться, что он является нейросетевым развитием идеи ab initio предсказания структуры белков. И снова мы возвращаемся к упомянутой идее — ни один из подходов предсказания структур не мертв, они лишь понемногу заимствуют все лучшее друг от друга.
Последующий прогресс
Помимо того, что Дэвид Бейкер не стеснялся использовать AlphaFold как метрику качества своей модели, Демис Хассабис и Джон Джампер также не стеснялись подсматривать идеи у Бейкера. Помимо самих белков, интересно ведь предсказывать и их комплексы с разными другими молекулами, такими как ДНК, гликаны, или, например, низкомолекулярные лиганды. В 2024 году на свет появилась AlphaFold3, в которой была добавлена диффузионная модель, способная подстраивать одни молекулы к другим и улучшать их геометрию в сомнительных или не очень точных местах. Не обошлось, конечно, без ложки дегтя — частенько данная модель начинала галлюцинировать и сшивать белки и другие молекулы в неожиданных местах, однако это явление совсем не снижает полезность данного инструмента.
Помимо непосредственно предсказания комплексов с другими молекулами, команда DeepMind, разрабатывающая AlphaFold, пошла дальше. В 2022 году AlphaFold2 был адаптирован под предсказания структур комплексов белков — AlphaFold-multimer. Достаточно ввести две или более последовательности белков, и программа предскажет их наиболее вероятный (с ее точки зрения) комплекс. Это действительно очень серьезная задача. Она позволяет исследовать механизмы сложных молекулярно-биологических явлений в клетках, когда два или более белков связываются в комплекс для совместного функционирования.
Но будто бы этого было мало. В 2024 году та же команда выпустила еще одну программу AlphaProteo. Ее основной целью стал de novo дизайн белковых структур, способных специфически связываться с заданными белками. А вот это уже совсем серьезная вещь. Подобные белки уже существуют, и исходя из их функции, похожей на функцию небезызвестных антител, их называют антителомиметиками. Однако антителомиметики часто имеют заранее выбранный каркас (например, DARPины используют в качестве основы повторы анкиринов и петли между ними для связывания других белков). Получаются антителомиметики с использованием направленной эволюции в таких сложных и дорогих методах, как фаговый дисплей. Из-за постоянства своих каркасов, не самого оптимального положения и состава петель в каждом отдельном примере, типичные антителомиметики показывают параметры связывания с мишенью хуже, чем антитела. AlphaProteo же не использует какой-то общий каркас, напротив, для каждого отдельного белка данная программа подбирает наиболее подходящую белковую структуру, которая с высокой вероятностью наоборот превзойдет антитела по своим характеристикам. У медицинской части читателей тут же встанет вопрос об иммуногенности получаемых структур, но эту сложную идею мы, пожалуй, оставим за скобками.
Что же дальше?
Мы с вами только что убедились, что выбор нобелевского комитета вполне себе достойный. Но что же нас ждет дальше?
Конечно, Бейкер не закончит на достигнутом и будет дальше совершенствовать свои алгоритмы. Сегодня базы данных экспериментально разрешенных структур белков пополняются все большим количеством данных криоэлектронной микроскопии, которая лишена проблем отклонения структуры от нативного состояния и игнорирования сложных участков рентгеновской кристаллографии, а также меньшей точности ядерного магнитного резонанса. Стало быть, дообучение перечисленных моделей на новых данных приведет к увеличению их точности, а также расширению сферы применения. То же самое касается Демиса Хассабиса и Джона Джампера. На базе достижений команды DeepMind под руководством их босса Alphabet Inc. (ранее Google) была сформирована компания Isomorphic Labs, которая занимается применением ИИ в биологии и медицине, а именно ускорением разработки новых лекарств, диагностических подходов и исследования сложных молекулярно-биологических механизмов.
Честно отвечая на вопрос «решена ли проблема фолдинга белка?», не получится однозначно ответить «да». Но использование технологий, разработанных и внедренных нобелевскими лауреатами по химии 2024 года, существенно продвинуло человечество в ее решении, а также создало абсолютно новые инструменты для прикладных задач.
Но давайте не будем забывать, что помимо белков существуют и другие важные молекулы, такие как ДНК, РНК, углеводы и др. В них таятся не менее сложные, важные и, что уж там, красивые механизмы. До сих пор не решена проблема фолдинга одноцепочечных нуклеиновых кислот, моделирования сложных гликанов на поверхностях белков, посттрансляционных модификаций, надежного предсказания комплексов разных молекул. Люди, о которых мы сегодня говорили, уже вступили на путь решения и этих проблем, однако их достижения пока далеки от идеала, и будет чрезвычайно интересно следить за тем, кто победит в других, не менее увлекательных гонках.


