
Муковисцидоз был впервые описан в 1938 году ученой и врачом Дороти Андерсен. Это смертельно опасное заболевание, наблюдаемое у новорожденных и детей, характеризующееся фиброзом поджелудочной железы с образованием кист, а также бронхоэктазов в легких и выделениями густой консистенции, которые закупоривают дыхательные пути. [1]. Впоследствии его классифицировали как генерализованную экзокринопатию, поражающую многие формы эпителиальных тканей — в печени, кишечнике, репродуктивном тракте и потовых железах [2]. В последующие десятилетия паллиативные методы лечения перешли в разряд стандартных и способствовали лишь продлению жизни, однако патогенный механизм оставался загадкой.
Важной вехой стало обнаружение в 1989 году «причинного» гена CFTR, который кодирует регулятор трансмембранной проводимости при муковисцидозе (CFTR) [3], хотя было неясно, приведет ли это открытие к усовершенствованию методов лечения. Со временем, Майклу Уэлшу, Хесусу Гонсалесу и Полу Негулеску удалось прояснить патогенез заболевания, детально исследовать фармакологические аспекты применительно к муковисцидозу и разработать революционную терапию этого заболевания. Их достижения отмечены Премией Ласкера-ДеБейки за клинические медицинские исследования 2025 года.
Еще до открытия CFTR Пол Квинтон из Калифорнийского университета получил ранние результаты, которые помогли Майклу Уэлшу с соавт. из Университета штата Айова показать, что дефектный белок при муковисцидозе регулирует анионную проницаемость клеток эпителия воздухоносных путей [4-6]. После описания CFTR [3] команде Уэлша удалось продемонстрировать, что рекомбинантный CFTR — несмотря на предсказанную структуру белка, которая не была похожа ни на один известный переносчик ионов — служил хлорным каналом [7]. Ученые по всему миру описывали сотни различных вариантов CFTR, связанных с муковисцидозом, однако мало что было известно о молекулярном механизме патогенеза. Уэлш возглавил усилия по определению и созданию системы классификации этих вариантов CFTR, основываясь на типе дисфункции CFTR (например, нарушение процессинга созревания, аномальное управление ионными каналами и преждевременный процессинг [8]) (рис. 1). Например, наиболее распространённый патологический вариант гена CFTR, приводящий к отсутствию одного остатка фенилаланина в аминокислотной позиции 508 (F508del) представляет собой дефект биогенеза, при котором неправильно свернутый белок CFTR разрушается клеточным механизмом “контроля качества” до того, как белок достигнет поверхности клетки. Уэлш с соавт. также показали, что рост клеток при низкой температуре или включение супрессорных мутаций второго сайта в ген CFTR компенсирует этот дефект [8,10]. Эти результаты заложили основу для исследования “малых молекул”, способных исправить F508del и другие нарушения свертывания белка CFTR.
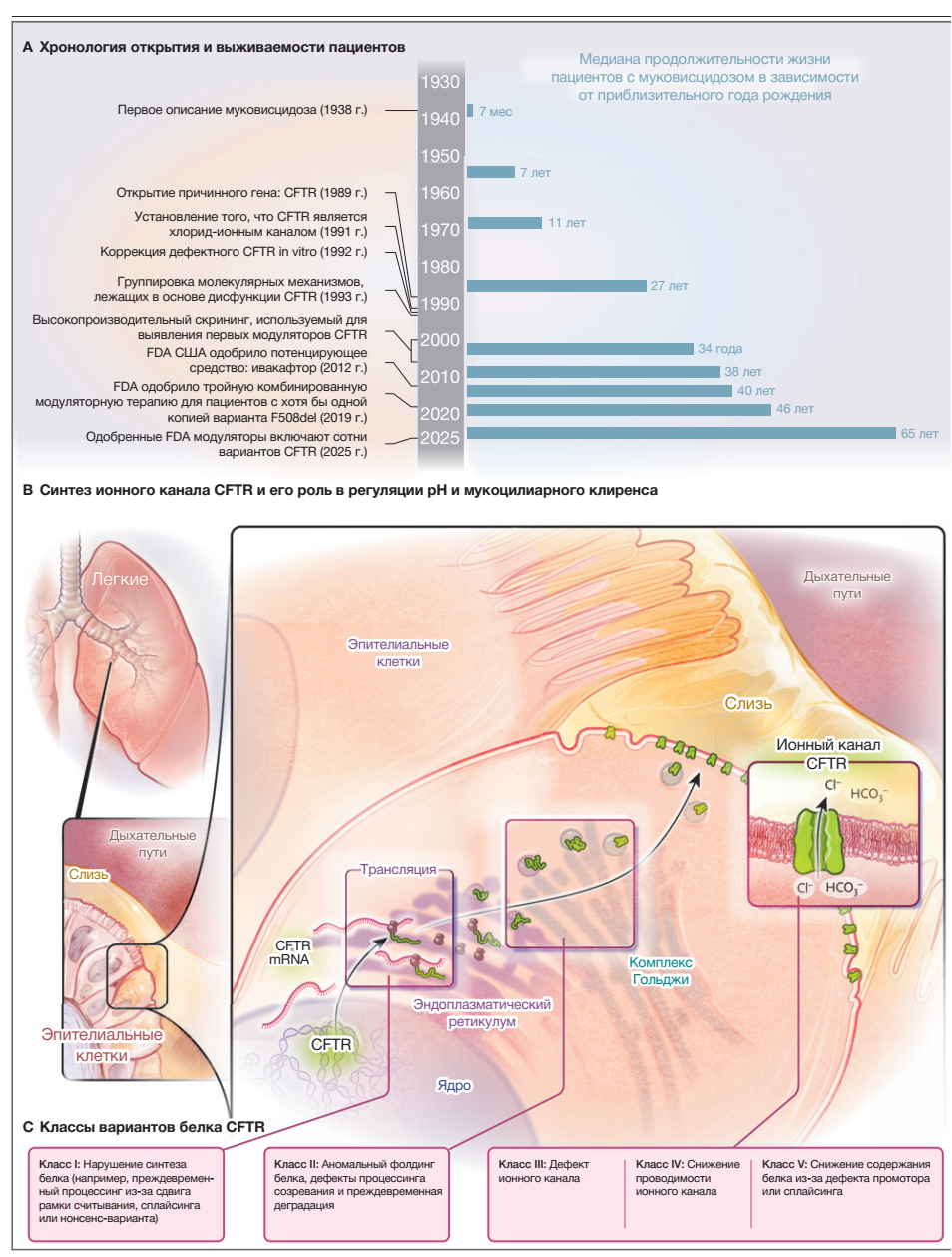
Рисунок 1. Достижения в изучении и лечении муковисцидоза.На панели A показана хронология, отражающая основные события, способствовавшие открытию препаратов-модуляторов для лечения муковисцидоза и медианную выживаемость пациентов с этим заболеванием. Данные о медианной выживаемости адаптированы из Davis Report [9] и Cystic Fibrosis Foundation 2024 Patient Registry Highlights Report (2024 Patient Registry Highlights | Cystic Fibrosis Foundation). Ряд паллиативных вмешательств, ставших доступными до 2012 года, служат частичным объяснением улучшения медианной выживаемости. Ожидается, что у пациентов, получающих новые модуляторы в качестве терапии, результаты будут более благоприятными, чем у тех, кто не получал такое лечение. На панели B показаны ключевые особенности, имеющие отношение к современному пониманию муковисцидоза. Регулятор трансмембранной проводимости при муковисцидозе (CFTR) представляет собой эпителиальный анионный канал, кодируемый геном CFTR. Канал CFTR регулирует состав и вязкость эпителиального и других экзокринных секретов дыхательных путей. В легких от CFTR зависят глубина слоя перицилиарной жидкости, образование подслизистого железистого секрета и мобилизация слизи дыхательных путей. На панели C показаны молекулярные механизмы, ответственные за дисфункцию CFTR. Известно, что сотни различных вариантов гена CFTR вызывают муковисцидоз. Подавляющее большинство из них можно сгруппировать в классы, как показано на рисунке. Также описаны дефекты VI класса, при которых нарушается встраивание, стабильность или рециркуляция белков-компонентов канала CFTR в плазматической мембране.
Научная необходимость поиска препаратов, способных восстанавливать мутировавший CFTR, представляла собой сложную задачу. Велся поиск малых молекул, специфически связывающих и восстанавливающих дефектную пептидную структуру CFTR, однако существование таких молекул было далеко не гарантировано. Лишь спустя десятилетия были получены высокоточные структурные данные о структуре белка CFTR и потребовалась совершенно новая стратегия. На сцену вышли ведущие ученые-фармакологи Пол Негулеску и Хесус Гонсалес, которые применили революционный метод скрининга библиотек соединений, что позволило быстро оценить сотни тысяч потенциальных препаратов для восстановления дефектного CFTR. В удивительном сочетании насущной терапевтической потребности и высокоспециализированных технологий Негулеску и Гонсалес (сначала в Aurora Biosciences, а затем в Vertex Pharmaceuticals) разработали персонализированный подход к разработке терапии дефектов CFTR. Стратегия была основана на многолетних фундаментальных научных исследованиях, финансируемых Национальными институтами здравоохранения (NIH), и ускорена финансовой поддержкой и руководством со стороны Фонда муковисцидоза (Cystic Fibrosis Foundation). Для совершения научного достижения, которое многие считали невозможным, были применены новые мембранные красители, чувствительные к потенциалу действия [11], передовые методы биофизического мониторинга и роботизированные системы автоматизации. Исследователям удалось обнаружить соединения, способные надежно связывать неправильно свернутый белок CFTR и преодолевать рефрактерные конформационные дефекты.
Для врачей-исследователей, проводивших ранние клинические испытания новых соединений-модуляторов CFTR, результаты можно было бы назвать просто чудесными. Первый модулятор CFTR, дошедший до стадии клинических испытаний, ивакафтор, действовал как потенцирующее средство, устраняя дефект ионного канала, вызванный мутацией G551D [2,12]. В течение десятилетий никакие терапевтические вмешательства не были способны снизить повышенный уровень хлоридов в поте (что является диагностическим признаком заболевания). В течение нескольких дней или недель после начала лечения ивакафтором уровень хлоридов в поте резко снизился, сопровождаясь очищением воздухоносных путей от мокроты и соответствующим улучшением функции легких [2,13]. Вслед за этим успехом последовало открытие таких малых молекул, как лумакафтор, элексакафтор, тезакафтор и ванзакафтор, которые корректируют аномальный биогенез (например, аномальный фолдинг белка, посттрансляционную модификацию и транспортировку на поверхность клетки) белка CFTR с мутацией F508del [2,14].
Было показано, что модуляторы вызывают неожиданную аллостерию; то есть препараты, разработанные для коррекции фолдинга белка у пациентов с определенным вариантом гена CTFR, зачастую также приносят большую пользу и для пациентов с другими вариантами. Для достижения максимальных клинических улучшений у пациентов с F508del требуется применение трех препаратов со взаимодополняющим действием на промежуточные звенья фолдинга белка CFTR или на конечную конформацию белка [2]. В настоящее время получены разрешения на применение отдельных препаратов или их комбинаций, направленных на более чем 250 вариантов CFTR, которые обуславливают фенотип муковисцидоза. Инновационные стандарты способствовали введению новых методов для расширения показаний к применению модуляторов CTFR. К примеру, использование модельных систем in vitro, которые прогнозируют улучшение дыхания у пациентов с вариантами гена CFTR, настолько редкими, что невозможно проведение клинических исследований III фазы[15].
Хотя значительное большинство людей с муковисцидозом в США теперь имеют доступ к высокоэффективным модуляторам, работа ученых и врачей далека от завершения. У части пациентов с этим заболеванием наблюдаются слабый ответ (или его отсутствие) на модуляторы, развиваются неприемлемые побочные эффекты при доступных схемах лечения, или же пациенты не могут позволить себе терапию, или являются носителями вариантов, которые вызывают преждевременную терминацию трансляции, аберрантный сплайсинг матричной РНК или другие дефекты, которые этими препаратами не исправляются. Исследователи муковисцидоза собираются ответить и на этот вызов, используя малые молекулы новейшего поколения, методы на основе нуклеотидов (например, редактирование генов и генный перенос CFTR) и другие стратегии. Достижения в исследовании и лечении муковисцидоза в прошлом стали символом гуманитарного блага, которого можно достичь, только когда вместе работают ученые-исследователи, фармацевтические компании, Национальные институты здравоохранения, благотворительные фонды, пациенты и их семьи. Действительно, когда Дороти Андерсен впервые описала это заболевание, зарегистрированная продолжительность жизни человека с муковисцидозом обычно составляла от нескольких месяцев до нескольких лет; на сегодняшний день предполагаемый медианный возраст выживания для пациентов с муковисцидозом, родившихся в период с 2020 по 2024 год и имеющих доступ к лечению, составляет 65 лет [16]. Доступные прогнозы предполагают большее улучшение здоровья и увеличение продолжительности жизни при начале приёма модуляторов с более раннего возраста. Новаторские достижения лауреатов премии Ласкера-Дебейки 2025 года дают все основания полагать, что лучшее ещё впереди.


