

Рецептор токсина сибирской язвы 1 (ANTXR1) — это интегрин-подобный трансмембранный белок. Он является платформой для связывания токсина сибирской язвы и опосредует взаимодействие клетки с внеклеточным матриксом. Новое исследование показывает, что ANTXR1 стабилизирует рецепторы трансформирующего фактора роста-β (TGFβ) на фибробластах сердца, что приводит к активации профибротической сигнализации и патологическому ремоделированию сердца.
Фиброз — это избыточное отложение внеклеточного матрикса (ВКМ), приводящее к рубцеванию ткани и дисфункции органов. Несмотря на то, что в сердце коллаген необходим для поддержания физиологической функции сердца, аномальная продукция ВКМ активированными кардиальными фибробластами связана с неблагоприятными клиническими исходами [1]. Переход фиброза из физиологического в патологическое состояние иллюстрируется ремоделированием сердца после инфаркта миокарда (ИМ), при котором нарушается тканевая перфузия, что приводит к массовой гибели кардиомиоцитов. Образование репаративного фиброзного рубца после ИМ имеет и положительную сторону, так как он формируется для предотвращения разрыва миокарда и сохранения его структурной целостности. Однако фиброзный рубец снижает сократимость миокарда и может привести к систолической дисфункции — как региональной, так и глобальной. Интерстициальное фиброзное ремоделирование сердца на фоне хронических коморбидных состояний, таких как артериальная гипертензия и ожирение, также ассоциировано с повышением пассивной жесткости миокарда и развитием диастолической дисфункции. Это способствует прогрессированию сердечной недостаточности с сохраненной фракцией выброса (ХСНсФВ). Фиброз может также нарушать проводимость миокарда, увеличивая риск развития аритмий. Несмотря на общепризнанную роль фиброза в патогенезе различных заболеваний сердца, в настоящее время отсутствует целенаправленная антифибротическая терапия. В данном выпуске Nature Cardiovascular Research Бокчелла и соавт. [2] раскрывают важнейшую функцию ANTXR1 в регуляции активации кардиальных фибробластов и фиброзного ремоделирования, указывая на новые потенциальные терапевтические стратегии для лечения сердечного фиброза.
Семейство ANTXR состоит из двух гомологичных интегрин-подобных трансмембранных белков — ANTXR1 (также называемого опухолевым эндотелиальным маркером 8, TEM8) и ANTXR2. ANTXR1 и ANTXR2 функционируют как рецепторы-мишени для токсина сибирской язвы (Bacillus anthracis), обеспечивая его проникновение в клетки [3]. В нормальных физиологических условиях рецепторы ANTXR экспрессируются на низком уровне в клетках различных типов и обеспечивают их взаимодействие с коллагеном внеклеточного матрикса (ECM) [4]. ANTXR1 был впервые идентифицирован как регулятор формирования сосудистой сети, связанной с опухолью. Кроме того, известно, что он высоко экспрессируется в ассоциированных с опухолью фибробластах [5]. Нокаут гена ANTXR1 у мышей приводит к развитию фиброза различных органов вследствие повышенной экспрессии белков внеклеточного матрикса фибробластами и снижения активности матриксных протеиназ [6]. На основании этих данных Бокчелла и соавт. выдвинули гипотезу, что у мышей с нокаутом ANTXR1 в ответ на факторы сердечного стресса будет развиваться более выраженный фиброз сердца.
Для проверки данной гипотезы авторы изучили экспрессию ANTXR1 в сердце. Уровни белка ANTXR1 были низкими в интактном миокарде, но значительно повышались в зоне инфаркта как в человеческом сердце, так и в мышином на фоне перенесенного ИМ. Аналогичным образом, уровень белка ANTXR1 был повышен в сердцах пациентов с дилатационной и гипертрофической кардиомиопатиями. Анализ данных РНК-секвенирования отдельных клеток (scRNA-seq) выявил увеличение количества транскриптов мРНК гена ANTXR1 в активированных кардиальных фибробластах. Однако, вопреки первоначальной гипотезе авторов, у мышей с полным нокаутом гена ANTXR1, подвергнутых модели ИМ путем постоянной окклюзии левой нисходящей коронарной артерии в течение 28 дней, объем инфаркта был меньше, а систолическая дисфункция — менее выраженной по сравнению с контрольной группой дикого типа, что может указывать скорее на профибротическую, а не антифибротическую роль данного рецептора в сердце.
Для оценки терапевтического потенциала воздействия на ANTXR1 авторы использовали межвидовые нейтрализующие антитела T8Ab, изначально разработанные для терапии онкологических заболеваний. T8Ab вводили внутрибрюшинно три раза в неделю в течение 6 недель, начиная спустя 24 часа после инфаркта миокарда, что имитировало реальную клиническую задержку лечения. У мышей, получавших антитела, наблюдалось улучшение сердечной функции и примерно две трети из них пережили постинфарктный период по сравнению лишь с одной третью в контрольной группе, при этом преимущества в выживаемости не отмечались у мышей с нокаутом ANTXR1, перенесших ИМ. В модели гипертонической болезни сердца у мышей, вызванной инфузией ангиотензина II (Ang II) и фенилэфрина (PE), T8Ab сохраняли как систолическую, так и диастолическую функцию, а также уменьшали интерстициальный фиброз. Даже когда терапия начиналась на фоне уже сниженной до 37 % фракции выброса (на 8-й день после начала введения Ang II/PE), T8Ab восстанавливали систолическую функцию и увеличивали продолжительность жизни. В модели ХСНсФВ, обусловленной ожирением и гипертензией, терапия T8Ab уменьшала фиброз, улучшала диастолические параметры и повышала толерантность к физической нагрузке, что непосредственно соответствует клинически значимым симптомам у пациентов. Между тем, делеция ANTXR1 в фибробластах заметно улучшала сердечную функцию как в моделях ИМ, так и Ang II/PE, причем гетерозиготные животные демонстрировали преимущества, сравнимые с таковыми при полном нокауте. Это позволяет предположить, что негативные эффекты ANTXR1 в фибробластах сердца проявляются после превышения порогового уровня экспрессии данного рецептора.
Последующий анализ методом scRNA-seq выявил, что в ответ на инфаркт миокарда или воздействие Ang II/PE, происходило появление ANTXR1-позитивных субпопуляций фибробластов с активированными профибротическими сигнальными путями, включающими трансформирующий фактор роста β (TGFβ) и белки, связанные с внеклеточным матриксом. Хотя транскрипты ANTXR1 обнаруживались широко, белок был сконцентрирован в тромбоспондин-4 (THBS4)-позитивных активированных фибробластах сердца, что указывает на уровень посттранскрипционного контроля, напоминающий таковой у других ассоциированных с фиброзом белков. Лечение T8Ab изменило экспрессию генов в фибробластах более существенно, чем в других типах клеток, включая шванновские, гладкомышечные и эпикардиальные клетки, определяя сердечные фибробласты как основные мишени действия ANTXR1. Удивительно, терапия T8Ab после инфаркта миокарда изменила экспрессию генов в фибробластах времязависимым образом. На 7-й день после инфаркта миокарда антитело усиливало экспрессию генов, кодирующих белки внеклеточного матрикса и матрично-клеточные белки, стабилизирующие рубцы, и снижало экспрессию токсичных факторов заболевания.
Напротив, к 14-му дню эти же программы были подавлены в результате лечения антителами T8Ab, а активированные при инфаркте миокарда пути, включая биосинтез коллагена и индукцию TGFβ, в значительной степени нормализовались. Дальнейшие исследования механизмов на культивированных фибробластах сердца показали прямую роль ANTXR1 в контроле профибротической сигнализации рецептора TGFβ (TGFβR). В фибробластах с нокаутом ANTXR1 или обработанных T8Ab уровни TGFβR1 были снижены, тогда как уровни TGFβR2 не изменились, и на основании ко-иммунопреципитационных исследований было установлено, что ANTXR1 и TGFβR1 формируют комплекс. Кроме того, делеция ANTXR1 или его нейтрализация с помощью T8Ab приводили к ослаблению TGFβ-опосредованной активации профибротических факторов транскрипции SMAD2 и YAP. В итоге авторы сформировали модель, в которой ингибирование ANTXR1 приводит к протеасомо-зависимой деградации TGFβR1, тем самым блокируя TGFβ-опосредованную профибротическую передачу сигналов и экспрессию генов в кардиальных фибробластах (рис. 1).
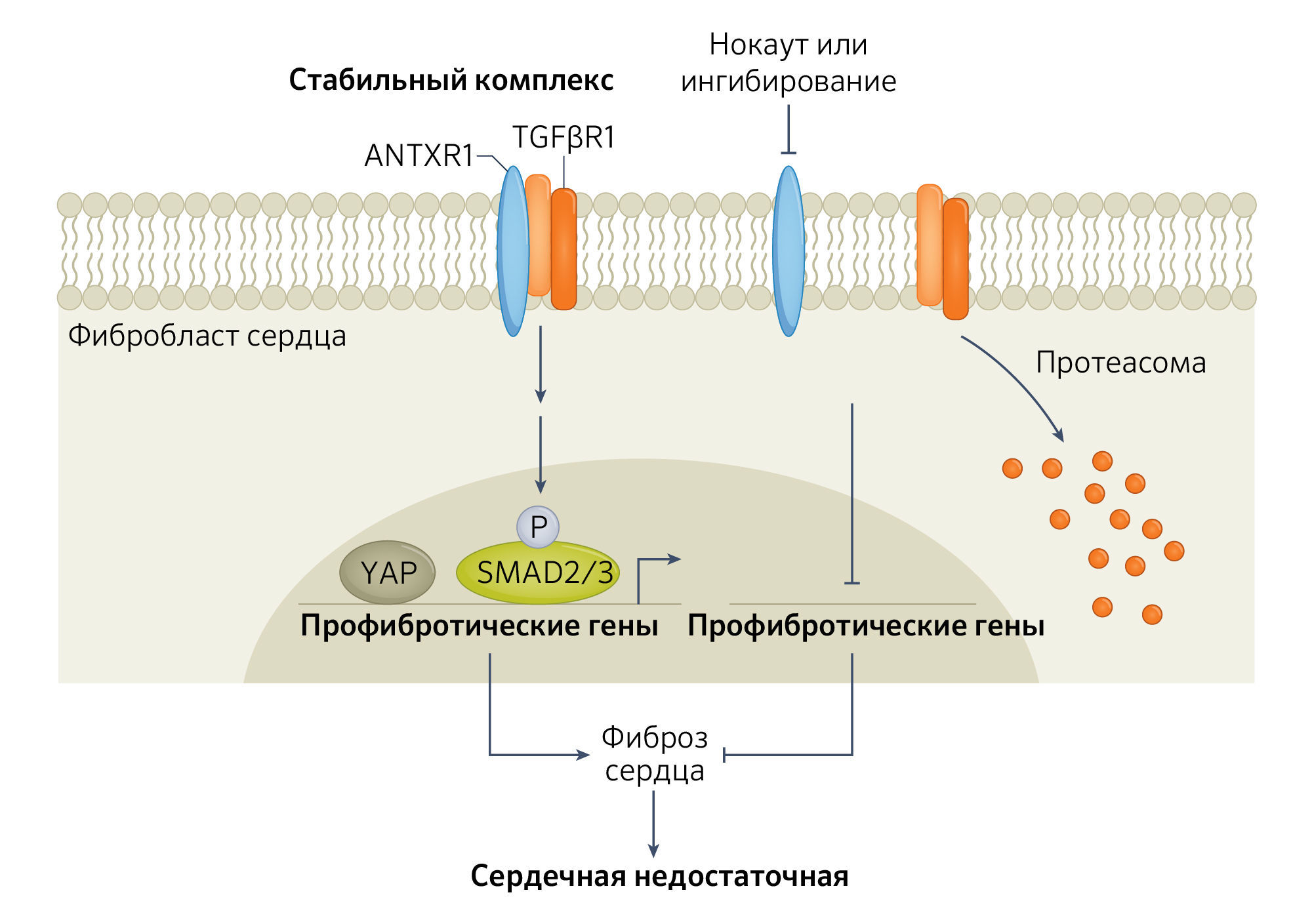
Рис.1. Модель регуляции фиброза сердца, опосредованного рецепторами антрактоксина.Antxr1 связывается с tgfbr1 и стабилизирует его, стимулируя передачу сигналов по нисходящему потоку, что приводит к экспрессии профибротических генов, опосредованных smad и yap, и фиброзу сердца. при ингибировании или делеции antxr1 протеасома разрушает tgfbr1, что приводит к подавлению экспрессии профибротического гена.
Проведенное Бокчеллой и соавт. исследование устанавливает ранее неизвестную роль ANTXR1 в контроле сердечного фиброза и предлагает подходы к воздействию на этот рецептор с целью коррекции фиброзного ремоделирования сердца. Однако, как и в случае любого новаторского открытия, многие вопросы остаются нерешенными. Например, ранее было показано, что ANTXR1 регулирует обмен коллагена [7], и в настоящей работе авторы предполагают, что этот обмен является ключевым фактором кардиофиброза, контролируемого ANTXR1 [2]. Концепция того, что деградация коллагена способствует кардиофиброзу, изначально кажется нелогичной, поскольку снижение уровня коллагена должно уменьшать размер рубца. Однако существуют данные, которые позволяют предположить, что повышение уровня коллагена в сердце может оказывать кардиопротективное действие [8]. Учитывая необычный и новаторский характер этой идеи, в будущих исследованиях следует использовать ортогональные методы анализа, например, маркировку стабильными изотопами и тяжелой водой [9], для количественной оценки влияния ингибирования ANTXR1 на коллагеновый обмен в сердце и других тканях.
С механистической точки зрения, тонкие различия в ответах кардиальных фибробластов и целого организма на лечение T8Ab по сравнению с нокаутом Antxr1 позволяют предположить, что эти два способа воздействия на рецептор вызывают перекрывающиеся, но частично различные эффекты. Например, в культивированных кардиальных фибробластах делеция ANTXR1 снижает их активацию, уменьшает экспрессию TGFβR1 и подавляет нисходящую передачу сигналов к SMAD и YAP. Эти эффекты существенно менее выражены в фибробластах сердца под воздействием T8Ab, тем не менее, нейтрализующее антитело было так же или даже более эффективно, чем нокаут ANTXR1, в улучшении сердечной функции in vivo. Более того, T8Ab снижало постинфарктную смертность у мышей, тогда как нокаут ANTXR1 не оказывал такого эффекта. Это открытие может быть связано со способностью T8Ab усиливать экспрессию генов внеклеточного матрикса через 7 дней после инфаркта миокарда, что позволяет предположить, что делеция ANTXR1 не приводит к реализации этой защитной программы, приводящей к образованию рубцов. Данную возможность следует проверить экспериментально, выполнив анализ scRNA-seq с сердцами мышей дикого типа и с нокаутом ANTXR1 на 7-й и 14-й день после инфаркта миокарда, аналогично тому, как это было сделано для мышей в эксперименте с T8Ab. Кроме того, необходимы дополнительные исследования для определения механизмов, посредством которых T8Ab усиливает профибротическую TGFβ-зависимую экспрессию генов на 7-й день после инфаркта миокарда, но подавляет этот же путь на 14-й день после инфаркта миокарда. Связаны ли эти противоположные эффекты с различным, зависящим от времени составом TGFβ-рецепторов в кардиальных фибробластах, который обуславливает различные ответы на T8Ab?
Результаты, полученные Бокчеллой и соавт., имеют важное значение для клинической интерпретации. Ведущей особенностью ANTXR1 как терапевтической мишени является его очень низкий уровень экспрессии в интактном сердце и повышение экспрессии в ответ на стресс, в том числе у человека. Таким образом, вмешательства, нацеленные на данный рецептор, должны быть «специфичными для заболевания» и минимально воздействовать на здоровые ткани. ANTXR1 представляет собой интегрин-подобный рецептор, а интегрины уже широко используются в качестве мишеней при разработке терапии фиброзных заболеваний, основанных на использовании блокирующих антител, малых молекул и пептидов [10].
На основании текущих результатов тестирование ингибирования ANTXR1 в контексте фиброза сердца у людей более чем оправдано. Но все еще предстоит определить, следует ли при этом использовать T8Ab, родственные нейтрализующие антитела или альтернативные подходы. Для определения приоритетов в подходах для клинических испытаний на людях может быть использована сравнительная оценка различных методов лечения, нацеленных на ANTXR1, на модели фиброза сердца у крупных животных. Что касается альтернативных вариантов ингибирования ANTXR1 in vivo, основываясь на знаниях, полученных из кристаллографических структур защитного антигена сибирской язвы в сочетании с ANTXR, возможно, удастся разработать небольшие молекулы и/или пептиды, которые смогут взаимодействовать с рецептором и ингибировать его [11, 12]. Кроме того, изучение механизмов регуляции экспрессии ANTXR1 (на транскрипционном, посттранскрипционном и посттрансляционном уровнях) может раскрыть уникальные подходы к воздействию на экспрессию данного рецептора в терапевтических целях. Наконец, учитывая его высокоиндуцируемую экспрессию в кардиальных фибробластах, ANTXR1 является перспективной мишенью для направления Т-клеток с химерным антигенным рецептором (CAR-T) к активированным кардиальным фибробластам с целью их элиминации, как было ранее описано для Т-клеток CAR, нацеленных на белок активации фибробластов-α (FAP) [13].
Сибирская язва (B. anthracis) впервые использовалась в качестве биологического оружия во время Первой мировой войны. Теперь, более 100 лет спустя, мы знаем, что воздействие на рецептор токсина, вырабатываемого этой смертоносной бактерией, может привести к разработке крайне необходимых антифибротических средств для лечения сердечно-сосудистых заболеваний.


